
Bild: ”Baby in the neonatal intensive care unit.” av Jacoplane (eller hans föräldrar i alla fall) – Eget arbete. Licens: CC BY-SA 3.0
- Introduktion
- Etiologi för pediatrisk konjugerad hyperbilirubinemi
- Diagnostik av konjugerad hyperbilirubinemi hos barn
- Historia
- Undersökning
- Laboratorietester vid pediatrisk konjugerad hyperbilirubinemi
- Kolestatiska orsaker till pediatrisk konjugerad hyperbilitubinemi
- Biliär atresi
- Koledokalcystor
- Geniska defekter vid pediatrisk konjugerad hyperbilirubinemi
- 1) Alagilles syndrom
- 2) Dubin-Johnsons syndrom
- Rotoriskt syndrom
Introduktion
Bilirubin är ett pigment (tetrapyrrol) som bildas i den normala kataboliska vägen från hemgruppen. Det är således en avfallsprodukt från destruktion av mogna röda blodkroppar. Enligt dess metabolism kan det klassificeras som konjugerat (korrelerat med direkt bilirubin) och okonjugerat bilirubin (korrelerat med indirekt bilirubin).
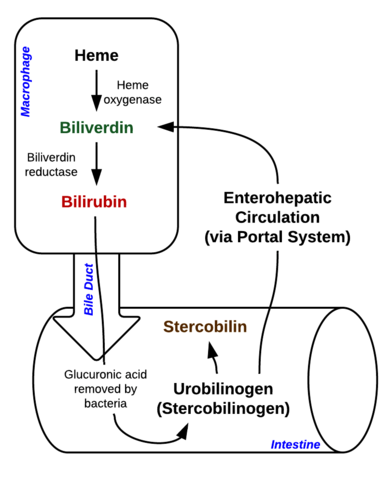
Bild: Bild: ”Breakdown of Heme in macrophages and intestine” av Johndheathcote. Licens: CC BY-SA 3.0
Hyperbilirubinemi kan definieras som en förhöjd koncentration av bilirubin i blodet, vilket tar sig uttryck i form av gulsot (även kallad ikterus). Gulsot uppträder när de gula bilirubinpigmenten deponeras i hud, sklera, slemhinnor och andra vävnader vilket leder till en gulaktig missfärgning.
Det normala intervallet för bilirubin är:
- Serum totalbilirubin mellan 0,2 och 1,2 mg/dL.
- Serum direktbilirubin mellan 0.1 och 0,4 mg/dL.
Det anses vara hyperbilirubinemi när:
- Direkt bilirubin i serum är ≥ 1 mg/dL (om totalt bilirubin är < 5 mg/dL).
- Konjugerat bilirubin i serum är > 20 % av totalt bilirubin (om totalt bilirubin är > 5 mg/dL).
Etiologi för pediatrisk konjugerad hyperbilirubinemi
I vissa nyfödda barn, särskilt för tidigt födda barn, kan en liten ökning av bilirubin betraktas som normal; den måste dock alltid undersökas eftersom en hög nivå av bilirubin på bekostnad av konjugerat bilirubin aldrig kan betraktas som normal.
Det finns många sjukdomar som kan orsaka hyperbilirubinemi, men de kan i stort sett klassificeras i två grupper: extrahepatiska sjukdomar (prehepatiska orsaker som hemolys på grund av inneboende defekter hos de röda blodkropparna eller extrinsiska orsaker som leder till att de röda blodkropparna spricker, eller posthepatiska orsaker som inkluderar obstruktion av gallgången) och intrahepatiska sjukdomar (leversjukdomar).
Det finns flera tillstånd av konjugerad hyperbilirubinemi hos barn :
- Infektioner: Virala (hepatit A-E, herpes, adenovirus, cytomegalovirus, HIV); bakteriella (sepsis, urinvägsinfektioner), röda hund, tuberkulos osv.
- Sjukdomar som orsakar obstruktion av gallgången, t.ex. biliär atresi, perforation av gallgången, kolelithiasis etc
- Sjukdomar i det endokrina systemet: hypotyreoidism, septooptisk dysplasi etc.
- Geniska/metabola sjukdomar: cystisk fibros, Alagilles syndrom, alfa-1-antitrypsin etc.
- Slagringsstörningar:
- Exponering för många läkemedel, inklusive ceftriaxon, metotrexat, erytromycin, tetracyklin osv.
Diagnostik av konjugerad hyperbilirubinemi hos barn
Historia
För att ställa diagnosen hyperbilirubinemi hos barn är det mycket viktigt att ta en bra klinisk historia, inklusive föräldrarnas och nära släktingars patologiska antecedenter, eventuell förekomst av släktskap mellan föräldrarna, eventuella komplikationer under graviditet och förlossning, eventuell exponering för en infektionssjukdom före eller efter födseln (t.ex. hepatit, malaria, cytomegalovirus och leptospiros) med mera.
Bild: Bild: ”Nyfödd spädbarn som genomgår fototerapi för att behandla neonatal gulsot” av Martin Pot. Licens: CC BY 3.0
Undersökning
Fysisk undersökning är mycket viktig vid diagnos av hyperbilirubinemi. Det viktigaste tecknet är gulsot eller gulaktig missfärgning av sklera, slemhinnor och hud. Barnets urin blir mörk och beroende på orsaken kan avföringen vara vit.
Det är också ytterst viktigt att utföra en neurologisk undersökning av barnet (reflexer och respons på yttre stimuli) för att fastställa affektionen i det centrala nervsystemet (som kallas kernicterus eller bilirubinencefalopati). Detta indikerar förekomsten av allvarlig hyperbilirubinemi.
Andra viktiga parametrar som måste undersökas är tillväxtparametrar, vitala tecken, hjärt- och andningsljud, bukundersökning (för att fastställa onormal tillväxt av mjälte eller lever).
Laboratorietester vid pediatrisk konjugerad hyperbilirubinemi
Laboratorietesterna som krävs är:
- Komplett blodkroppsräkning som är nödvändig för screening av hemolys.
- Serumnivåerna av aminotransferaser görs för att undersöka leverfunktionen. De omfattar aspartataminotransferas och alaninaminotransferas .
- Serologisk screening för virus.
- Alkalisk fosfatas eftersom en förhöjning av det kan tyda på obstruktion av gallgångarna.
- Gamma-glutamyltranspeptidas vars nivåer kan hjälpa till att skilja en hepatisk källa till det förhöjda ALP från andra orsaker.
- Fraktionerat bilirubin.
I andra termer är leverfunktionstester (LFT eller LF) nödvändiga för diagnosen och de omfattar protrombintid (PT/INR), aktiverad partiell tromboplastintid (aPTT), albumin, bilirubin (direkt och indirekt) och levertransaminaser (AST och ALT). Biopsi och bildundersökningar är vanligtvis reserverade för de fall med en oklar diagnos eller för att utesluta obstruktiva patologier.
Kolestatiska orsaker till pediatrisk konjugerad hyperbilitubinemi
Biliär atresi
Biliär atresi är en annan mycket frekvent orsak till konjugerad hyperbilirubinemi. Patienten kan ha en fullständig eller partiell obstruktion av det extrahepatiska gallträdet på olika ställen; de vanligaste delarna som drabbas är dock leverkanalen och den gemensamma gallgången. Partiell obstruktion orsakas vanligen av en fibroinflammation längs gallgången.
Denna fibroinflammation beror på en perinatal sjukdom, vanligen en virusinfektion som påverkar gallgångens slemhinna. När infektionen fortskrider reagerar det immunologiska systemet och det uppstår en epitelförtjockning i slemhinnan som leder till obstruktion av gallgången. Med tiden utvecklar den prolifererande slemhinnan skleros och fibros, vilket orsakar total kanalatresi under den första levnadsmånaden och dess efterföljande komplikationer.
I diagnosen av biliär atresi är det också mycket viktigt att ta en fullständig klinisk anamnes, utföra en bra fysisk undersökning och laboratorietester som tidigare nämnts; i det här fallet kan dock bildgivande tekniker (som ultraljud, hepatobiliär skanning (HIDA) eller till och med ett intraoperativt kolangiogram) vara mycket användbara för att diagnostisera sjukdomen.
Ingen primär medicinsk behandling är effektiv vid korrigering av biliär atresi; när diagnosen väl är bekräftad är därför ett kirurgiskt ingrepp nödvändigt och i många fall kan själva operationen vara diagnostisk. Det kan göras som ett intraoperativt kolangiogram eller en Kasai portoenterostomi. Levertransplantationer är reserverade för allvarliga fall.
Koledokalcystor
För det sista kan koledokalcystor, mer kända som medfödd gallgångsdilatation, också orsaka konjugerad hyperbilirubinemi. Den vanligaste medfödda abnormiteten vid koledokalcystor är utvidgningen av den extrahepatiska gallgången (1 av 100 000-150 000 nyfödda). Symptomen på koledokockcystor kan uppträda i vilken ålder som helst, men de ses karakteristiskt med obstruktiv gulsot tillsammans med buksmärta hos spädbarn och barn. De har en kvinnlig predilektion med ett förhållande mellan kvinnor och män på cirka 3-4:1. De är vanligare hos vissa asiatiska raser.
Typer av koledokalcystor: Ursprungligen beskrevs tre typer av koledokala cystor, men Todani och medarbetare klassificerade dessa ytterligare i fem typer som beskrivs nedan:
- Typ I: Det finns en dilatation av den gemensamma gallgången. Den kan vara cystisk, fokal eller fusiform (ses i 90-95 % av fallen).
- Typ II: Här ses ett divertikel i den extrahepatiska gallgången.
- Typ III: Kännetecknas av koledokoceler.
- Typer IV: Det finns två typer, typ IV-A och typ IV-B. Typ IV-A är den näst vanligaste typen och definieras som både intrahepatisk och extrahepatisk dilatation av gallträdet. Typ IV-B innebär den sällsynta missbildningen av flera extrahepatiska cystor.
- Typ V: Denna typ kallas även Carolis sjukdom när den är förknippad med hepatisk fibros. Typ V innefattar enstaka eller flera intrahepatiska cystor.
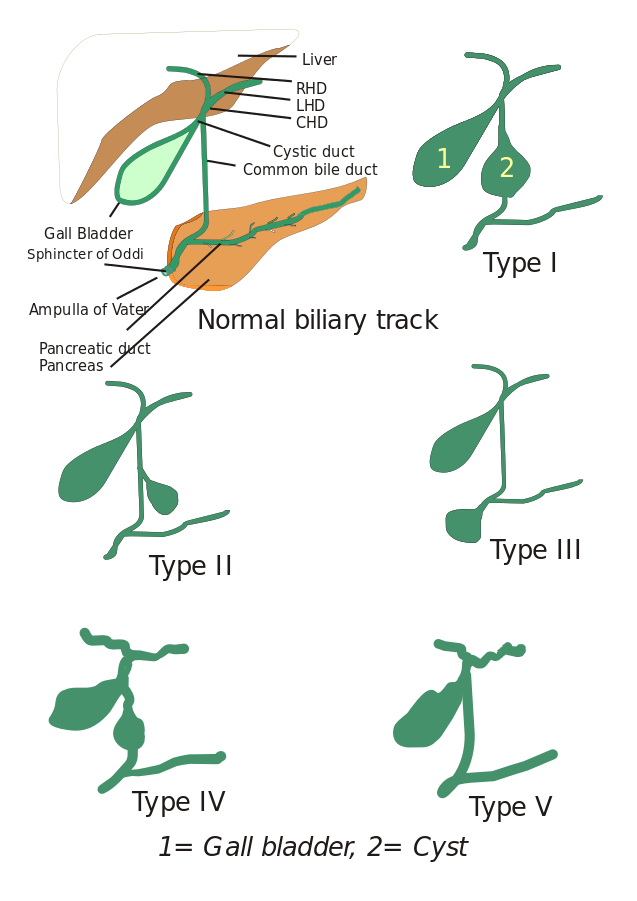
Bild: ”5 typer av koledokala cystor. Typ I: Dilatation av extrahepatisk gallgång, typ II: Cysta från gemensam gallgång (CBD), typ III: Choledochocele eller dilatation av distala delen av CBD, typ IV: Dilatation av både extrahepatisk och intrahepatisk gallgång, typ V: Carolis sjukdom, dilatation av endast den intrahepatiska kanalen. CHD: Common hepatical duct, LHD: Vänster hepatisk ductus och RHD: Högra hepatiska kanalen: LHD: LHD, höger hepatiska kanal och höger hepatiska kanal. Genom I, Drriad. Licens: CC BY-SA 3.0
Det är vanligt hos spädbarn i åldern 1-3 månader. Kliniskt presenterar de sig med feber, smärta i höger övre kvadrant av buken, acholisk eller blek avföring och hepatomegali och denna bild liknar biliär atresi. Vid prenatal diagnos av koledokalcystor är gulsot inte uppenbar förrän 1-3 veckor efter födseln. När koledokockcystan uppträder senare blir den inte kliniskt uppenbar förrän efter två års ålder och kan uppträda med en klassisk triad av buksmärta, palpabel bukmassa och gulsot, varav två förekommer hos nästan 85 % av barnen vid presentationstillfället. Andra vanliga kännetecken är kolangit, pankreatit och gallperitonit på grund av cystans ruptur.
Laboratoriska undersökningar samt bilddiagnostik är nödvändiga för att ställa en definitiv diagnos. Laboratorieundersökningar omfattar de konventionella leverfunktionstesterna, CBC och koagulationsprofiler. Bildundersökningar omfattar bland annat ultraljudsscanning, datortomografi, magnetresonanstomografi, magnetresonanskolangiopankreatografi och endoskopisk retrograd kolangiopankreatografi.
Ultraljudsscanning underlättar diagnosen av koledokockcystor och har en mycket hög specificitet på 97 % hos barn, men låg diagnostisk noggrannhet antenatalt; därför används den som primär diagnostisk bilddiagnostik vid neonatal gulsot som kvarstår mer än två veckor efter födseln. Den är också användbar vid differentiering mellan koledokockcystor och biliär atresi.
Användningen av datortomografi för att diagnostisera koledokockcystor är kontroversiell. Små cystor eller koledochocele missas på datortomografi men ses lätt på magnetresonanskolangiopankreatografi (MRCP). CT-skanning kan vara bättre än MRCP under den postoperativa perioden för att upptäcka läget för biliär-enterisk anastomos och för att definiera eventuell stenos av denna.
Magnetisk resonanskolangiopankreatografi (MRCP) är överlägsen när det gäller att upptäcka och definiera lesioner. Numera betraktas den som guldstandard vid avbildning av koledokalcystor; den har dock den nackdelen att den inte exakt kan upptäcka en eventuell avvikande pankreatisk-biliär förening. Detta spelar dock ingen större roll när det gäller att bestämma patientens behandling. Dessutom är MRCP:s känslighet större hos vuxna än hos spädbarn.
Endoskopisk retrograd kolangiopankreatografi (ERCP) är det bästa verktyget för att känna till den biliära anatomin och är därmed till hjälp vid diagnostik av koledokockcystor.
Behandlingen innefattar i första hand kirurgiska ingrepp. Ett internt dräneringsförfarande som kallas cyst-enterostomi (som kan vara antingen cyst-duodenektomi eller cyst-jejunostomi) gjordes tidigare för kirurgisk behandling av koledokala cystor; olika komplikationer sågs dock postoperativt såsom malignitet i den kvarvarande cystan, pankreatit och kolangit; därför övergavs de.
För närvarande är det rekommenderade kirurgiska ingreppet excision av cystan och därefter Roux-en-Y hepaticojejunostomi eller koledochojejunostomi. På grund av cystexcisionen minskar förekomsten av strikturbildning postoperativt. Andra alternativ, dvs. hepatisk duodenotomi, har föreslagits så att anastomosen är tillgänglig för ERCP i händelse av postoperativa komplikationer. Användningen av hepatisk duodenotomi har dock inte accepterats i stor utsträckning eftersom den kan orsaka biliär reflux och kolangit.
Efter cystborttagning är sondering och riklig sköljning av de intrahepatiska kanalerna med koksaltlösning nödvändig för att helt avlägsna slam och eventuella stenar från kanalsystemet. Dessutom kan obstruktionen finnas i det proximala gallsystemet som kan dilateras. Följaktligen är en intraoperativ kolangiografi obligatorisk före excision av cystan.
Geniska defekter vid pediatrisk konjugerad hyperbilirubinemi
Det finns många ärftliga syndrom som är relaterade till hyperbilirubinemi och intrahepatisk kolestas. Många av dem är relaterade till genetiska mutationer inklusive genen SERPINAI (alfa 1- antitrypsin), JAG1 (orsakar Alagilles syndrom), ATP8B1 (även känd som FIC1), ABCB11 (gallsaltsexportpump ), MDR3 (ABCB4) och MRP2 (orsakar Dubin-Johnsons syndrom).
1) Alagilles syndrom
Alagilles syndrom är en autosomalt dominant genetisk sjukdom som kan påverka många delar av kroppen. Mutationen förekommer i den korta armen av kromosom 20 (20p12). En av 20 eller 30 mutationer uppstår de novo. Ett av de mest drabbade organen vid Alagilles syndrom är levern och gallgångarna. Dessa missbildningar av gallgångarna orsakar gallans ansamling i levern, dvs. kolestasier, som orsakar ärrbildning i levern och därmed förändrar dess funktion.
De tecken och symtom som uppstår till följd av leverskador vid Alagilles syndrom kan omfatta gulsot, kliande hud och xanthomer. Hjärtat påverkas dock också och patienten kan ha lungstenos. Vid pulmonisk stenos finns det ett försämrat blodflöde från hjärtat till lungorna. Det kan också finnas medföljande medfödda hjärtsjukdomar, såsom ventrikelseptumdefekter, tetralogi av Fallot osv. Hjärnan, ryggmärgen, njurarna och blodkärlen kan också påverkas.
Alagilles syndrom har karakteristiska ansiktsdrag. Patienten har vanligtvis ett triangulärt ansikte, en bred och framträdande panna, bred näsbrygga, djupt liggande ögon och en liten, spetsig haka. Diagnosen är svår och kräver minst tre av de typiska fysiska dragen, bevis på obstruktion av gallgången (kolestas) och en leverbiopsi.
Behandlingen är beroende av sjukdomens svårighetsgrad vid Alagilles syndrom. Vid lindrig sjukdom ges ursodeoxikolsyra för att underlätta gallflödet och antihistaminer som difenhydramin ges för att kontrollera pruritus. Vid allvarliga fall kan en levertransplantation krävas. Vitamintillskott har visat sig vara till hjälp, särskilt med vitamin A, D, E och K. Zinktillskott är också fördelaktigt. Dessa patienter har en oförmåga att absorbera dessa vitaminer och därför bidrar tillskott till att underlätta optimal tillväxt hos patienten.
2) Dubin-Johnsons syndrom
Detta syndrom kännetecknas av isolerad konjugerad kronisk hyperbilirubinemi, utan tecken på hemolys på grund av en defekt i elimineringen av konjugerat bilirubin i gallan (intrahepatisk kolestas). Den orsakas av en mutation i MRP2-genen. Det är ett godartat tillstånd som inte kräver särskild behandling.
Rotoriskt syndrom
Det är en sällsynt sjukdom som kännetecknas av kronisk konjugerad och okonjugerad hyperbilirubinemi utan hemolys. Det genetiska arvet är fortfarande inte känt. Den uppstår på grund av en defekt i leverens lagring av konjugerat bilirubin, som läcker ut i plasman och orsakar hyperbilirubinemi.
Studiera för läkarutbildning och nämnder med Lecturio.
- USMLE Steg 1
- USMLE Steg 2
- COMLEX Level 1
- COMLEX Level 2
- ENARM
- NEET