
Billede: “Baby in the neonatal intensive care unit.” af Jacoplane (eller i hvert fald hans forældre) – Eget værk. Licens: CC BY-SA 3.0
- Indledning
- Ætiologi af pædiatrisk konjugeret hyperbilirubinæmi
- Diagnose af konjugeret hyperbilirubinæmi hos børn
- Historie
- Undersøgelse
- Laboratoriske undersøgelser af konjugeret hyperbilirubinæmi hos børn
- Kolestatiske årsager til konjugeret hyperbilitubinæmi hos børn
- Bilær atresi
- Coledochalcyster
- Genetiske defekter ved pædiatrisk konjugeret hyperbilirubinæmi
- 1) Alagille syndrom
- 2) Dubin-Johnson syndrom
- Rotorsyndrom
Indledning
Bilirubin er et pigment (tetrapyrrol), der dannes i den normale kataboliske vej fra hæmgruppen. Det er således et affaldsprodukt fra modne røde blodlegemers destruktion. I henhold til dets metabolisme kan det klassificeres som konjugeret (korreleret med direkte bilirubin) og ukonjugeret bilirubin (korreleret med indirekte bilirubin).
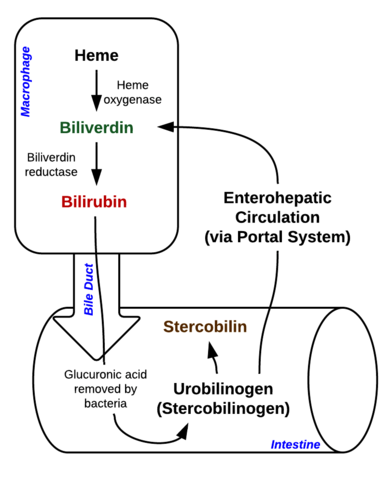
Billede: Billede: “Breakdown of Heme in macrophages and intestine” af Johndheathcote. Licens: CC BY-SA 3.0
Hyperbilirubinæmi kan defineres som en forhøjet koncentration af bilirubin i blodet, hvilket kommer til udtryk i form af gulsot (også kaldet icterus). Gulsot opstår, når de gule bilirubinpigmenter aflejres i hud, sclera, slimhinder og andre væv, hvilket fører til gullig misfarvning.
Det normale bilirubininterval er:
- Serum totalbilirubin mellem 0,2 og 1,2 mg/dL.
- Serum direkte bilirubin mellem 0.1 og 0,4 mg/dL.
Det betragtes som hyperbilirubinæmi, når:
- Serum direkte bilirubin er ≥ 1 mg/dL (hvis totalbilirubin er < 5mg/dL).
- Serum konjugeret bilirubin er > 20 % af totalbilirubin (hvis totalbilirubin er > 5mg/dL).
Ætiologi af pædiatrisk konjugeret hyperbilirubinæmi
I nogle nyfødte børn, især for tidligt fødte børn, kan en lille stigning i bilirubin betragtes som normal; det skal dog altid undersøges, fordi et højt niveau af bilirubin på bekostning af konjugeret bilirubin aldrig kan betragtes som normalt.
Der er mange sygdomme, der kan forårsage hyperbilirubinæmi, men de kan groft sagt inddeles i to grupper: ekstrahepatiske lidelser (præhepatiske årsager som hæmolyse på grund af intrinsiske defekter i de røde blodlegemer eller extrinsiske årsager, der fører til bristning af røde blodlegemer, eller posthepatiske årsager, herunder obstruktion af galdepassagen) og intrahepatiske lidelser (leversygdomme).
Der er flere tilstande med konjugeret hyperbilirubinæmi hos børn :
- Infektioner: Virale (hepatitis A-E, herpes, adenovirus, cytomegalovirus, HIV); bakterielle (sepsis, urinvejsinfektioner), røde hunde, tuberkulose osv.
- Sygdomme, der forårsager obstruktion af galdegangen, såsom galdeatresi, perforation af galdegangen, cholelithiasis osv
- Sygdomme i det endokrine system: hypothyroididsm, septooptisk dysplasi osv.
- Genetisk/metabolisk: cystisk fibrose, Alagilles syndrom, alfa-1-antitrypsin osv.
- Lagringsforstyrrelser:
- Besiddelse af mange lægemidler, herunder ceftriaxon, methotrexat, erythromycin, tetracyclin osv.
Diagnose af konjugeret hyperbilirubinæmi hos børn
Historie
For at stille en diagnose af hyperbilirubinæmi hos børn er det meget vigtigt at tage en god klinisk historie, herunder forældrenes og nære slægtninges patologiske antecedenter, den mulige eksistens af slægtskab mellem forældrene, mulige komplikationer under graviditet og fødsel, mulig eksponering for en smitsom sygdom før eller efter fødslen (såsom hepatitis, malaria, cytomegalovirus og leptospirose) blandt andre.
Billede: Billede: “Nyfødt spædbarn under lysbehandling til behandling af neonatal gulsot” af Martin Pot. Licens: CC BY 3.0
Undersøgelse
Fysisk undersøgelse er meget vigtig i forbindelse med diagnosticering af hyperbilirubinæmi. Det vigtigste tegn er gulsot eller gullig misfarvning af sclera, slimhinder og hud. Barnets urin bliver mørk, og afhængig af årsagen kan afføringen være hvid.
Det er også yderst vigtigt at foretage en neurologisk undersøgelse af barnet (reflekser og respons på ydre stimuli) for at bestemme lidelsen i centralnervesystemet (der er kendt som kernicterus eller bilirubin-encefalopati). Dette indikerer tilstedeværelsen af alvorlig hyperbilirubinæmi.
Andre vigtige parametre, der skal undersøges, omfatter vækstparametre, vitale tegn, hjerte- og åndedrætslyde, abdominalundersøgelse (for at bestemme unormal vækst af milt eller lever).
Laboratoriske undersøgelser af konjugeret hyperbilirubinæmi hos børn
De nødvendige laboratorieundersøgelser er:
- Fuldstændig blodcelletælling, som er nødvendig for screening af hæmolyse.
- Serum aminotransferase-niveauer foretages for at undersøge leverens funktion. De omfatter aspartataminotransferase og alaninaminotransferase.
- Serologisk screening for virus.
- Alkalisk fosfatase, da en stigning i den kan indikere en obstruktion af galdegangene.
- Gamma-glutamyltranspeptidase, hvis niveauer kan hjælpe med at skelne en hepatisk kilde til den forhøjede ALP fra andre årsager.
- Fraktioneret bilirubin.
I andre tilfælde er leverfunktionstest (LFT eller LF) nødvendige for diagnosen, og de omfatter protrombintid (PT/INR), aktiveret partiel tromboplastintid (aPTT), albumin, bilirubin (direkte og indirekte) og levertransaminaser (AST og ALT). Biopsi og billeddannende undersøgelser er normalt forbeholdt de tilfælde med en uklar diagnose eller udelukke obstruktive patologier.
Kolestatiske årsager til konjugeret hyperbilitubinæmi hos børn
Bilær atresi
Bilær atresi er en anden meget hyppig årsag til konjugeret hyperbilirubinæmi. Patienten kan have en fuldstændig eller delvis obstruktion af det ekstrahepatiske galdetræ på forskellige steder; de mest almindelige dele, der rammes, er dog leverkanalen og den almindelige galdegang. Den delvise obstruktion skyldes normalt en fibro-inflammation langs galdegangen.
Denne fibro-inflammation skyldes en perinatal sygdom, normalt en virusinfektion, der påvirker galdegangens slimhinde. Når infektionen skrider frem, reagerer det immunologiske system, og der sker en epithelial fortykkelse af slimhinden, hvilket fører til obstruktion af galdegangen. Efterhånden som tiden skrider frem, udvikler den prolifererende slimhinde sklerose og fibrose, hvilket forårsager total kanalatresi i løbet af den første måned af livet og de efterfølgende komplikationer.
I diagnosen af biliær atresi er det også meget vigtigt at tage en komplet klinisk anamnese, udføre en god fysisk undersøgelse og laboratorieprøver som nævnt tidligere; i dette tilfælde kan billeddannelsesteknikker (som ultralyd, hepatobiliær scanning (HIDA) eller endog et intraoperativt kolangiogram) dog være meget nyttige i forbindelse med diagnosticering af sygdommen.
Ingen primær medicinsk behandling er effektiv til korrektion af biliær atresi; når diagnosen er bekræftet, er det derfor nødvendigt med et kirurgisk indgreb, og i mange tilfælde kan selve operationen være diagnostisk. Det kan ske som et intraoperativt kolangiogram eller en Kasai-portoenterostomi. Levertransplantationer er forbeholdt svære tilfælde.
Coledochalcyster
Endeligt kan choledochalcyster, bedre kendt som medfødt dilatation af galdegangene, også forårsage konjugeret hyperbilirubinæmi. Den mest almindelige medfødte abnormitet ved koledochalcyster er udvidelsen af den ekstrahepatiske galdegang (1 ud af hver 100.000-150.000 nyfødte). Symptomerne på koledochale cyster kan forekomme i alle aldre; de ses dog karakteristisk med obstruktiv gulsot sammen med mavesmerter hos spædbørn og børn. De har en kvindelig forkærlighed med et forhold mellem kvinder og mænd på ca. 3-4:1. De er mere almindelige hos visse asiatiske racer.
Typer af koledochale cyster: Oprindeligt blev der beskrevet tre typer af choledochalcyster, men Todani og medarbejdere har yderligere klassificeret disse i fem typer, som er beskrevet nedenfor:
- Type I: Der er dilatation af den almindelige galdegang. Den kan være cystisk, fokal eller fusiform (ses i 90-95% af tilfældene).
- Type II: Her ses et divertikel af den ekstrahepatiske galdegang.
- Type III: Karakteriseret ved koledochoceles.
- Typer IV: Der er to typer, type IV-A og type IV-B. Type IV-A er den næsthyppigste type og defineres som både intrahepatisk og ekstrahepatisk dilatation af biliærtræet. Type IV-B omfatter den sjældne misdannelse af flere ekstrahepatiske cyster.
- Type V: Denne type er også kendt som Carolis sygdom, når den er forbundet med hepatisk fibrose. Type V omfatter enkelte eller flere intrahepatiske cyster.
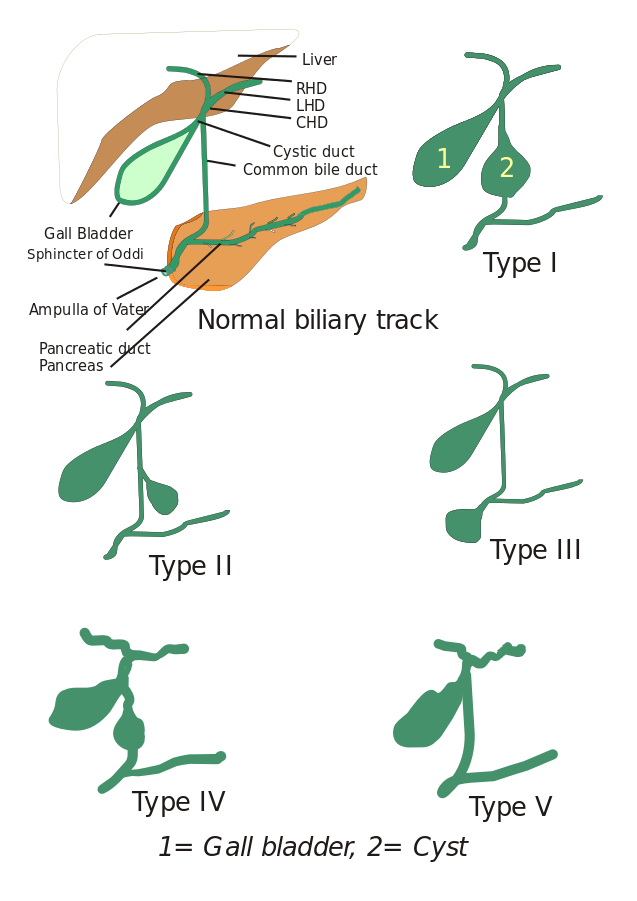
Billede: “5 typer af choledochale cyster. Type I: Dilatation af den ekstrahepatiske galdegang; Type II: Cyste fra den almindelige galdegang (CBD); Type III: Choledochocele eller dilatation af den distale del af CBD, type IV: Dilatation af både den ekstrahepatiske og intrahepatiske galdegang; Type V: Caroli-sygdom, dilatation af kun den intrahepatiske kanal. CHD: Fælles hepatisk ductus, LHD: venstre hepatisk ductus og RHD: højre hepatisk ductus.” Af I, Drriad. Licens: CC BY-SA 3.0
Det ses almindeligvis hos spædbørn i alderen 1 til 3 måneder. Klinisk præsenterer de sig med feber, smerter i højre øvre kvadrant af abdomen, acholisk eller bleg afføring og hepatomegali, og dette billede ligner biliær atresi. I tilfælde af prænatal diagnose af koledochalcyste er gulsot ikke synlig før 1 til 3 uger efter fødslen. Når koledochalcysten viser sig senere, bliver den ikke klinisk tydelig før efter 2-årsalderen og kan vise sig med en klassisk triade af abdominale smerter, palpabel abdominal masse og gulsot, hvoraf to findes hos næsten 85 % af børnene på det tidspunkt, hvor de præsenteres. Andre almindelige træk er kolangitis, pancreatitis og galdeperitonitis som følge af bristning af cysten.
Laboratoriske undersøgelser samt billeddannelse er nødvendige for at stille en endelig diagnose. Laboratorieundersøgelser omfatter de konventionelle leverfunktionstest, CBC og koagulationsprofiler. Billeddannende undersøgelser omfatter bl.a. ultralydsscanning, computertomografi, magnetisk resonansbilleder, magnetisk resonans-kolangiopancreatografi og endoskopisk retrograd kolangiopancreatografi.
Ultralydsscanning hjælper med at diagnosticere koledochalcyster, og den har en meget høj specificitet på 97 % hos børn, men en lav diagnostisk nøjagtighed antenatalt; den anvendes derfor som primær diagnostisk billeddannelsesmodalitet ved neonatal gulsot, der varer mere end 2 uger efter fødslen. Den er også nyttig til differentiering mellem koledochalcyster og biliær atresi.
Brug af computertomografiske scanninger til diagnosticering af koledochalcyster er kontroversiel. Cyster af lille størrelse eller koledochocele overses på CT-scanninger, men ses let på magnetisk resonans-kolangiopancreatografi (MRCP). CT-scanning kan være bedre end MRCP i den postoperative periode til at påvise beliggenheden af den biliær-enteriske anastomose og til at definere en eventuel stenose af denne.
Magnetisk resonans-kolangiopancreatografi (MRCP) er bedre til at påvise og definere læsioner. I dag betragtes den som guldstandard i forbindelse med billeddannelse af koledochalcyster; den har dog den ulempe, at den ikke præcist kan påvise enhver anomal pankreatiskobiliær forening. Ikke desto mindre spiller dette ikke nogen større rolle ved fastlæggelsen af patientens behandling. Desuden er MRCP’s følsomhed større hos voksne end hos spædbørn.
Endoskopisk retrograd kolangiopancreatografi (ERCP) er det bedste redskab til at kende den galdeformede anatomi og er således nyttig ved diagnosticering af koledochalcyster.
Behandlingen omfatter først og fremmest kirurgisk indgreb. En intern dræningsprocedure kendt som en cyst-enterostomi (der enten kunne være cyst-duodenektomi eller cyst-jejunostomi) blev tidligere udført til kirurgisk behandling af koledochale cyster; der blev dog set forskellige komplikationer postoperativt såsom malignitet i den tilbageværende cyste, pancreatitis og kolangitis; de blev derfor opgivet.
På nuværende tidspunkt er den anbefalede kirurgiske procedure cysterekcision og derefter Roux-en-Y hepaticojejunostomi eller koledochojejunostomi. På grund af cysteskæring er der en reduktion i forekomsten af strikturdannelse postoperativt. Andre alternativer, f.eks. hepatisk duodenotomi, er blevet foreslået, således at anastomosen er tilgængelig for ERCP i tilfælde af postoperative komplikationer. Brugen af hepatisk duodenotomi er imidlertid ikke blevet bredt accepteret, da den kan forårsage galdeflux og kolangitis.
Efter cysteskæring er sondering og rigelig skylning af de intrahepatiske kanaler med saltvand nødvendig for at fjerne slam og eventuelle sten fuldstændigt fra kanalsystemet. Endvidere kan obstruktionen være til stede i det proximale galdesystem, som kan udvides. Derfor er en intraoperativ kolangiografi obligatorisk inden excision af cysten.
Genetiske defekter ved pædiatrisk konjugeret hyperbilirubinæmi
Der findes mange arvelige syndromer i forbindelse med hyperbilirubinæmi og intrahepatisk kolestase. Mange af dem er relateret til genetiske mutationer, herunder genet SERPINAI (alfa 1- antitrypsin), JAG1 (forårsager Alagille syndrom), ATP8B1 (også kendt som FIC1), ABCB11 (galdesalt eksportpumpe ), MDR3 (ABCB4) og MRP2 (forårsager Dubin-Johnson syndrom).
1) Alagille syndrom
Alagille syndrom er en autosomal dominant genetisk lidelse, der kan påvirke mange dele af kroppen. Mutationen forekommer i den korte arm af kromosom 20 (20p12). En ud af hver 20. eller 30. mutation opstår de novo. Et af de mest berørte organer ved Alagilles syndrom er leveren og galdegangene. Disse galdegangsmisdannelser forårsager ophobning af galde i leveren, dvs. kolestase, hvilket medfører arvævsdannelse i leveren og derved ændrer dens funktion.
Tegnene og symptomerne som følge af leverskader ved Alagilles syndrom kan omfatte gulsot, kløende hud og xanthomer. Hjertet er dog også påvirket, og patienten kan have pulmonal stenose. Ved pulmonalstenose er der nedsat blodgennemstrømning fra hjertet til lungerne. Der kan også være medfølgende medfødte hjertesygdomme som f.eks. ventrikelseptumdefekter, tetralogi af Fallot osv. Hjernen, rygmarven, nyrerne og blodkarrene kan også være påvirket.
Alagille syndrom har karakteristiske ansigtstræk. Patienten har normalt et trekantet ansigt, en bred og fremtrædende pande, en bred næseryg, dybtliggende øjne og en lille, spids hage. Diagnosen er vanskelig og kræver mindst 3 af de typiske fysiske træk, tegn på obstruktion af galdegangene (kolestase) og en leverbiopsi.
Behandlingen afhænger af sygdommens sværhedsgrad ved Alagilles syndrom. Ved mild sygdom gives ursodeoxycholsyre for at lette galdeflowet, og antihistaminer som diphenhydramin gives for at kontrollere pruritus. I svære tilfælde kan det være nødvendigt med en levertransplantation. Vitamintilskud har vist sig at være nyttigt, især med vitaminerne A, D, E og K. Zinktilskud er også gavnligt. Disse patienter har en manglende evne til at optage disse vitaminer, og derfor hjælper tilskud til at lette patientens optimale vækst.
2) Dubin-Johnson syndrom
Dette syndrom er karakteriseret ved isoleret konjugeret kronisk hyperbilirubinæmi uden tegn på hæmolyse på grund af en defekt i elimineringen af konjugeret bilirubin i galde (intrahepatisk kolestase). Den er forårsaget af en mutation i MRP2-genet. Det er en godartet tilstand, der ikke kræver specifik behandling.
Rotorsyndrom
Det er en sjælden lidelse, der er karakteriseret ved kronisk konjugeret og ukonjugeret hyperbilirubinæmi uden hæmolyse. Den genetiske arv er stadig ikke kendt. Den opstår på grund af en defekt i leverens lagring af konjugeret bilirubin, som lækker ud i plasmaet og forårsager hyperbilirubinæmi.
Lær til medicinstudier og bestyrelser med Lecturio.
- USMLE Step 1
- USMLE Step 2
- COMLEX Level 1
- COMLEX Level 2
- ENARM
- NEET