
Imagen: «Bebé en la unidad de cuidados intensivos neonatales» por Jacoplane (bueno, sus padres al menos) – Obra propia. Licencia: CC BY-SA 3.0
- Introducción
- Etiología de la hiperbilirrubinemia conjugada pediátrica
- Diagnóstico de la hiperbilirrubinemia conjugada pediátrica
- Historia
- Examen
- Pruebas de laboratorio de la hiperbilirrubinemia conjugada pediátrica
- Causas colestásicas de hiperbilitubinemia conjugada pediátrica
- Atresia biliar
- Quistes coledocianos
- Defectos genéticos de la hiperbilirrubinemia conjugada pediátrica
- 1) Síndrome de Alagille
- 2) Síndrome de Dubin-Johnson
- Síndrome de Rotor
Introducción
La bilirrubina es un pigmento (tetrapirrol) que se forma en la vía catabólica normal a partir del grupo hemo. Es, por tanto, un producto de desecho de la destrucción de los glóbulos rojos maduros. Según su metabolismo, puede clasificarse en bilirrubina conjugada (correlativa a la bilirrubina directa) y bilirrubina no conjugada (correlativa a la bilirrubina indirecta).
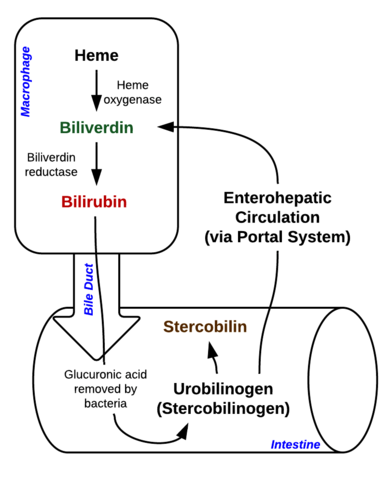
Imagen: «Descomposición del hemo en macrófagos e intestino» por Johndheathcote. Licencia: CC BY-SA 3.0
La hiperbilirrubinemia puede definirse como una concentración elevada de bilirrubina en la sangre, que se expresa en forma de ictericia (también llamada ictericia). La ictericia aparece cuando los pigmentos amarillos de la bilirrubina se depositan en la piel, la esclerótica, las membranas mucosas y otros tejidos dando lugar a una decoloración amarillenta.
El rango normal de bilirrubina es:
- Bilirrubina total sérica entre 0,2 y 1,2 mg/dL.
- Bilirrubina directa sérica entre 0.1 y 0,4 mg/dL.
Se considera hiperbilirrubinemia cuando:
- La bilirrubina directa sérica es ≥ 1 mg/dL (si la bilirrubina total es < 5mg/dL).
- La bilirrubina conjugada sérica es > 20% de la bilirrubina total (si la bilirrubina total es > 5mg/dL).
Etiología de la hiperbilirrubinemia conjugada pediátrica
En algunos recién nacidos, especialmente los prematuros, un ligero aumento de la bilirrubina puede considerarse normal; sin embargo, siempre debe estudiarse porque un nivel elevado de bilirrubina a expensas de la bilirrubina conjugada nunca puede considerarse normal.
Hay muchas enfermedades que pueden causar hiperbilirrubinemia, pero pueden clasificarse a grandes rasgos en dos grupos: trastornos extrahepáticos (causas prehepáticas como la hemólisis debida a defectos intrínsecos de los glóbulos rojos o causas extrínsecas que conducen a la ruptura de los glóbulos rojos, o causas poshepáticas que incluyen la obstrucción del paso biliar) y trastornos intrahepáticos (enfermedades del hígado).
Hay varias condiciones de hiperbilirrubinemia conjugada en niños :
- Infecciones: Virales (hepatitis A-E, herpes, adenovirus, citomegalovirus, VIH); bacterianas (sepsis, infecciones urinarias), rubéola, tuberculosis, etc.
- Trastornos que causan obstrucción de la vía biliar como atresia biliar, perforación de la vía biliar, colelitiasis, etc
- Enfermedades del sistema endocrino: hipotiroidismo, displasia septoóptica, etc.
- Genética/metabólica: fibrosis quística, síndrome de Alagille, alfa-1-antitripsina, etc.
- Trastornos del almacenamiento: Síndrome de Gaucher, enfermedades de almacenamiento de glucógeno, Nieman-Pick, etc.
- Exposición a muchos medicamentos incluyendo ceftriaxona, metotrexato, eritromicina, tetraciclina, etc.
Diagnóstico de la hiperbilirrubinemia conjugada pediátrica
Historia
Para realizar el diagnóstico de la hiperbilirrubinemia en niños, es muy importante realizar una buena historia clínica, que incluya los antecedentes patológicos de los padres y familiares cercanos, la posible existencia de consanguinidad entre los padres, las posibles complicaciones durante el embarazo y el parto, la posible exposición a una enfermedad infecciosa antes o después del nacimiento (como la hepatitis, la malaria, el citomegalovirus y la leptospirosis) entre otras.
Imagen: «Recién nacido sometido a fototerapia para tratar la ictericia neonatal» por Martin Pot. Licencia: CC BY 3.0
Examen
La exploración física es muy importante en el diagnóstico de la hiperbilirrubinemia. El signo principal es la ictericia o coloración amarillenta de la esclerótica, las mucosas y la piel. La orina del bebé se vuelve oscura y, dependiendo de la causa, las heces pueden ser blancas.
También es muy importante realizar un examen neurológico del bebé (reflejos y respuesta a estímulos externos) para determinar la afección del sistema nervioso central (que se conoce como kernicterus o encefalopatía bilirrubínica). Esto indica la presencia de una hiperbilirrubinemia grave.
Otros parámetros importantes que deben examinarse son los parámetros de crecimiento, los signos vitales, los soplos cardíacos y respiratorios, el examen abdominal (para determinar el crecimiento anormal del bazo o del hígado).
Pruebas de laboratorio de la hiperbilirrubinemia conjugada pediátrica
Las pruebas de laboratorio necesarias son:
- Cuento completo de células sanguíneas que es necesario para el cribado de la hemólisis.
- Los niveles de aminotransferasas séricas se realizan para estudiar la función del hígado. Incluyen la aspartato aminotransferasa y la alanina aminotransferasa.
- Control serológico de los virus.
- Fosfatasa alcalina ya que un aumento de la misma puede indicar obstrucción de las vías biliares.
- Gamma-glutamil transpeptidasa cuyos niveles pueden ayudar a diferenciar un origen hepático de la ALP elevada de otras causas.
- Bilirrubina fraccionada.
En otros términos, las pruebas de función hepática (LFTs o LFs) son necesarias para el diagnóstico e incluyen el tiempo de protrombina (PT/INR), el tiempo de tromboplastina parcial activado (aPTT), la albúmina, la bilirrubina (directa e indirecta) y las transaminasas hepáticas (AST y ALT). La biopsia y los estudios de imagen suelen reservarse para aquellos casos con un diagnóstico poco claro o descartar patologías obstructivas.
Causas colestásicas de hiperbilitubinemia conjugada pediátrica
Atresia biliar
La atresia biliar es otra causa muy frecuente de hiperbilirrubinemia conjugada. El paciente puede tener una obstrucción completa o parcial del árbol biliar extrahepático en diferentes puntos; sin embargo, las partes más comúnmente afectadas son el conducto hepático y el conducto biliar común. La obstrucción parcial suele estar causada por una fibroinflamación a lo largo del conducto biliar.
Esta fibroinflamación se debe a una enfermedad perinatal, normalmente una infección vírica que afecta a la mucosa del conducto biliar. Cuando la infección progresa, el sistema inmunológico responde y se produce un engrosamiento epitelial en la mucosa que conduce a la obstrucción del conducto. A medida que avanza el tiempo, la mucosa proliferante desarrolla esclerosis y fibrosis, provocando la atresia total del conducto durante el primer mes de vida y sus complicaciones posteriores.
En el diagnóstico de la atresia biliar, también es muy importante realizar una historia clínica completa, una buena exploración física y pruebas de laboratorio como se ha mencionado anteriormente; sin embargo, en este caso, las técnicas de imagen (como la ecografía, la gammagrafía hepatobiliar (HIDA) o incluso un colangiograma intraoperatorio) pueden ser muy útiles en el diagnóstico de la enfermedad.
Ningún tratamiento médico primario es eficaz en la corrección de la atresia biliar; por ello, una vez confirmado el diagnóstico, es necesaria la intervención quirúrgica y, en muchos casos, la propia cirugía puede ser diagnóstica. Puede realizarse una colangiografía intraoperatoria o una portoenterostomía de Kasai. Los trasplantes de hígado se reservan para los casos graves.
Quistes coledocianos
Por último, los quistes coledocianos, más conocidos como dilatación congénita de la vía biliar, también pueden causar hiperbilirrubinemia conjugada. La anomalía congénita más común de los quistes coledocianos es el agrandamiento del conducto biliar extrahepático (1 de cada 100.000-150.000 recién nacidos). Los síntomas de los quistes coledocianos pueden presentarse a cualquier edad; sin embargo, es característico verlos con ictericia obstructiva junto con dolor abdominal en bebés y niños. Tienen una predilección por las mujeres, con una proporción de mujeres a hombres de aproximadamente 3-4:1. Son más comunes en ciertas razas asiáticas.
Tipos de quistes coledocianos: Inicialmente, se describieron tres tipos de quistes coledocianos, pero Todani y sus colaboradores los clasificaron en cinco tipos que se describen a continuación:
- Tipo I: Hay dilatación del conducto biliar común. Puede ser quística, focal o fusiforme (se observa en el 90-95% de los casos).
- Tipo II: Aquí se observa un divertículo del conducto biliar extrahepático.
- Tipo III: Se caracteriza por coledoceles.
- Tipos IV: Es de dos tipos, Tipo IV-A y Tipo IV-B. El tipo IV-A es el segundo más frecuente y se define como una dilatación tanto intra como extrahepática del árbol biliar. El tipo IV-B implica la rara malformación de múltiples quistes extrahepáticos.
- Tipo V: Este tipo también se conoce como enfermedad de Caroli cuando se asocia con fibrosis hepática. El tipo V incluye quistes intrahepáticos únicos o múltiples.
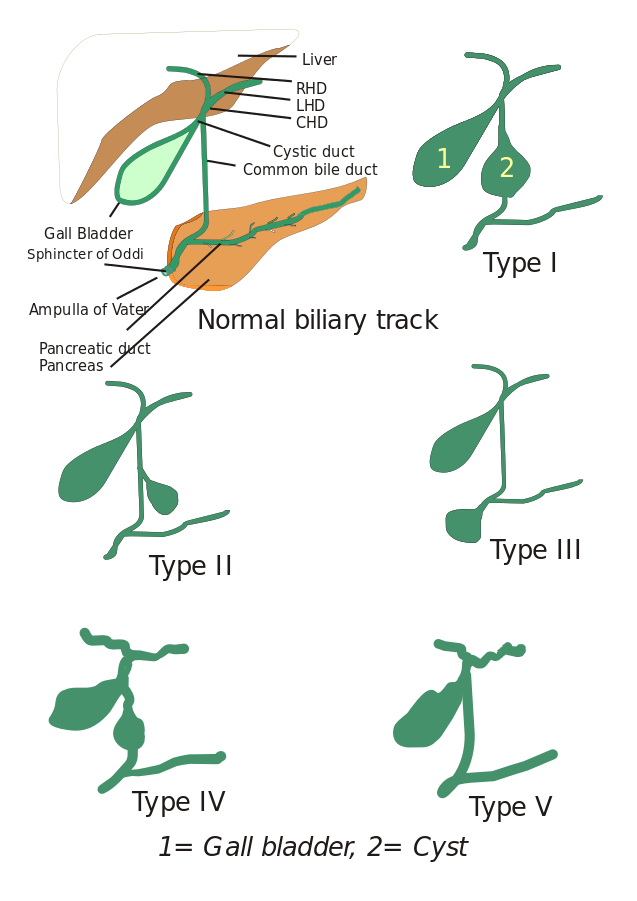
Imagen: «5 tipos de quistes coledocianos. Tipo I: Dilatación del conducto biliar extrahepático; Tipo II: Quiste del conducto biliar común (CBD); Tipo III: Coledocele o dilatación de la parte distal del CBD, Tipo IV: Dilatación del conducto extrahepático e intrahepático; Tipo V: Enfermedad de Caroli, Dilatación del conducto intrahepático solamente. CHD: Conducto hepático común, LHD: Conducto hepático izquierdo y RHD: Conducto hepático derecho». Por I, Drriad. Licencia: CC BY-SA 3.0
Se observa con frecuencia en lactantes de 1 a 3 meses. Clínicamente se presentan con fiebre, dolor en el cuadrante superior derecho del abdomen, heces acólicas o pálidas y hepatomegalia y este cuadro es similar a la atresia biliar. En el caso del diagnóstico prenatal del quiste coledociano, la ictericia no se manifiesta hasta 1 ó 3 semanas después del nacimiento. Cuando el quiste coledociano se presenta más tarde, no se hace clínicamente evidente hasta después de los 2 años de edad y puede presentarse con una tríada clásica de dolor abdominal, masa abdominal palpable e ictericia; de las cuales dos se encuentran en casi el 85% de los niños en el momento de la presentación. Otras características comunes son colangitis, pancreatitis y peritonitis biliar debido a la rotura del quiste.
Las investigaciones de laboratorio, así como las imágenes, son necesarias para un diagnóstico definitivo. Las investigaciones de laboratorio incluyen las pruebas convencionales de las funciones hepáticas, el hemograma y los perfiles de coagulación. Los estudios de imagen incluyen la ecografía, la tomografía computarizada, la resonancia magnética, la colangiopancreatografía por resonancia magnética y la colangiopancreatografía retrógrada endoscópica, entre otros.
La ecografía ayuda en el diagnóstico de los quistes coledocianos y tiene una especificidad muy alta del 97% en niños, pero una precisión diagnóstica baja prenatalmente; por lo tanto, se utiliza como modalidad de imagen diagnóstica primaria para la ictericia neonatal que persiste más de 2 semanas después del nacimiento. También es útil para diferenciar los quistes coledocianos de la atresia biliar.
El uso de la tomografía computarizada para el diagnóstico de los quistes coledocianos es controvertido. Los quistes de pequeño tamaño o el coledocele no se detectan en la TC, pero se ven fácilmente en la colangiopancreatografía por resonancia magnética (CPRM). La TC puede ser mejor que la CPRM en el periodo postoperatorio para detectar la localización de la anastomosis biliar-entérica y para definir cualquier estenosis de la misma.
La colangiopancreatografía por resonancia magnética (CPRM) es superior para detectar y definir las lesiones. Hoy en día, se considera el estándar de oro en la imagen de los quistes coledocianos; sin embargo, tiene el inconveniente de no poder detectar con precisión ninguna unificación pancreaticobiliar anómala. No obstante, esto no desempeña un papel importante a la hora de determinar el tratamiento de los pacientes. Además, la sensibilidad de la CPRM es mayor en los adultos que en los lactantes.
La colangiopancreatografía retrógrada endoscópica (CPRE) es la mejor herramienta para conocer la anatomía biliar y, por tanto, es útil en el diagnóstico de los quistes coledocianos.
El tratamiento incluye principalmente la intervención quirúrgica. En el pasado se realizaba un procedimiento de drenaje interno conocido como quiste-enterostomía (que podía ser quiste-duodenectomía o quiste-yeyunostomía) para el tratamiento quirúrgico de los quistes coledocianos; sin embargo, se observaron diversas complicaciones en el postoperatorio como malignidad en el quiste restante, pancreatitis y colangitis; por tanto, se abandonaron.
En la actualidad, el procedimiento quirúrgico recomendado es la escisión del quiste y luego la hepaticoyeyunostomía en Y de Roux o la coledocoyeyunostomía. Debido a la escisión del quiste, se reduce la incidencia de formación de estenosis en el postoperatorio. Se han sugerido otras alternativas, como la duodenotomía hepática, para que la anastomosis sea accesible a la CPRE en caso de complicaciones postoperatorias. Sin embargo, el uso de la duodenotomía hepática no ha sido ampliamente aceptado ya que puede causar reflujo biliar y colangitis.
Después de la extirpación del quiste, es necesario sondear y realizar un lavado abundante de los conductos intrahepáticos con solución salina para eliminar completamente el lodo y los posibles cálculos del sistema ductal. Además, la obstrucción puede estar presente en el sistema biliar proximal que puede ser dilatado. En consecuencia, antes de la escisión del quiste, es obligatorio realizar una colangiografía intraoperatoria.
Defectos genéticos de la hiperbilirrubinemia conjugada pediátrica
Hay una gran cantidad de síndromes hereditarios relacionados con la hiperbilirrubinemia y la colestasis intrahepática. Muchos de ellos están relacionados con mutaciones genéticas que incluyen el gen SERPINAI (alfa 1 antitripsina), JAG1 (causante del síndrome de Alagille), ATP8B1 (también conocido como FIC1), ABCB11 (bomba de exportación de sales biliares ), MDR3 (ABCB4) y MRP2 (causante del síndrome de Dubin-Johnson).
1) Síndrome de Alagille
El síndrome de Alagille es un trastorno genético autosómico dominante que puede afectar a muchas partes del cuerpo. La mutación se produce en el brazo corto del cromosoma 20 (20p12). Una de cada 20 o 30 mutaciones se produce de novo. Uno de los órganos más afectados en el síndrome de Alagille es el hígado y los conductos biliares. Estas malformaciones de los conductos biliares provocan una acumulación de bilis en el hígado, es decir, colestasis, que causa cicatrices en el hígado, alterando así su funcionamiento.
Los signos y síntomas derivados del daño hepático en el síndrome de Alagille pueden incluir ictericia, picor en la piel y xantomas. Sin embargo, el corazón también se ve afectado y el paciente puede presentar estenosis pulmonar. En la estenosis pulmonar, el flujo sanguíneo desde el corazón hacia los pulmones se ve afectado. También puede haber cardiopatías congénitas coexistentes, como defectos del tabique ventricular, tetralogía de Fallot, etc. El cerebro, la médula espinal, los riñones y los vasos sanguíneos también pueden estar afectados.
El síndrome de Alagille tiene unos rasgos faciales característicos. El paciente suele tener una cara triangular, una frente ancha y prominente, un puente nasal ancho, ojos hundidos y un mentón pequeño y puntiagudo. El diagnóstico es difícil y requiere al menos 3 de los rasgos físicos típicos, la evidencia de obstrucción del conducto biliar (colestasis) y una biopsia de hígado.
El tratamiento depende de la gravedad de la enfermedad en el síndrome de Alagille. En el caso de la enfermedad leve, se administra ácido ursodesoxicólico para facilitar el flujo biliar y antihistamínicos como la difenhidramina para controlar el prurito. En los casos graves, puede ser necesario un trasplante de hígado. Se ha comprobado que la administración de suplementos vitamínicos es útil, especialmente de vitaminas A, D, E y K. La administración de suplementos de zinc también es beneficiosa. Estos pacientes tienen una incapacidad para absorber estas vitaminas y, por tanto, la suplementación ayuda a facilitar el crecimiento óptimo del paciente.
2) Síndrome de Dubin-Johnson
Este síndrome se caracteriza por una hiperbilirrubinemia crónica conjugada aislada, sin evidencia de hemólisis debido a un defecto en la eliminación de la bilirrubina conjugada en la bilis (colestasis intrahepática). Está causada por una mutación en el gen MRP2. Es una enfermedad benigna que no requiere tratamiento específico.
Síndrome de Rotor
Es un trastorno raro caracterizado por hiperbilirrubinemia crónica conjugada y no conjugada sin hemólisis. La herencia genética aún no se conoce. Se produce por un defecto de almacenamiento de la bilirrubina conjugada en el hígado, que se filtra al plasma causando hiperbilirrubinemia.
Estudia para la facultad de medicina y los exámenes con Lecturio.
- USMLE Paso 1
- USMLE Paso 2
- COMLEX Nivel 1
- COMLEX Nivel 2
- ENARMAR
- NEET