
Image: “Bebé na unidade de cuidados intensivos neonatais” por Jacoplane (bem, os pais dele de qualquer maneira) – Trabalho próprio. Licença: CC BY-SA 3.0
- Introdução
- Etiologia da Hiperbilirrubinemia Conjugada Pediátrica
- Diagnóstico de Hiperbilirrubinemia Conjugada Pediátrica
- História
- Exame
- Testes laboratoriais de hiperbilirrubinemia conjugada pediátrica
- Causas da hiperbilirrubinemia conjugada pediátrica
- Atresia biliar
- Cistos Coledochal
- Defeitos Genéticos da Hiperbilirrubinemia Conjugada Pediátrica
- 1) Síndrome de Alagille
- 2) Síndrome de Dubin-Johnson
- Síndrome Motora
Introdução
Bilirrubina é um pigmento (tetrapyrrole) que é formado na via catabólica normal do grupo heme. É, portanto, um produto residual de destruição de glóbulos vermelhos maduros. De acordo com seu metabolismo, pode ser classificado como conjugado (correlacionado com bilirrubina direta) e não conjugado (correlacionado com bilirrubina indireta).
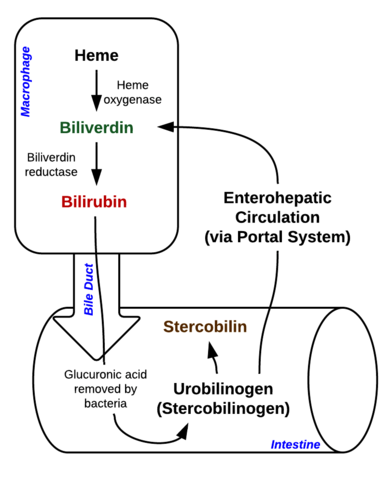
Image: “Breakdown of Heme in macrophages and intestine” por Johndheathcote. Licença: CC BY-SA 3.0
Hiperbilirrubinemia pode ser definida como uma concentração elevada de bilirrubina no sangue, que é expressa na forma de icterícia (também chamada icterícia). A icterícia aparece quando os pigmentos amarelos da bilirrubina são depositados na pele, esclera, membranas mucosas e outros tecidos levando a uma descoloração amarelada.
A faixa normal da bilirrubina é:
- Soro bilirrubina total entre 0,2 e 1,2 mg/dL.
- Soro bilirrubina direta entre 0.1 e 0,4 mg/dL.
É considerado como hiperbilirrubinemia quando:
- Soro bilirrubina directa é ≥ 1 mg/dL (se bilirrubina total for < 5mg/dL).
- Soro bilirrubina conjugada é > 20% da bilirrubina total (se bilirrubina total for > 5mg/dL).
Etiologia da Hiperbilirrubinemia Conjugada Pediátrica
Em alguns recém-nascidos, especialmente prematuros, um ligeiro aumento da bilirrubina pode ser considerado normal; no entanto, deve ser sempre estudado porque um alto nível de bilirrubina às custas da bilirrubina conjugada nunca pode ser considerado normal.
Existem muitas doenças que podem causar hiperbilirrubinemia, mas podem ser amplamente classificadas em dois grupos: distúrbios extra-hepáticos (causas pré-hepáticas como hemólise devido a defeitos intrínsecos dos glóbulos vermelhos ou causas extrínsecas levando à ruptura dos glóbulos vermelhos, ou causas pós-hepáticas incluindo obstrução da passagem biliar) e distúrbios intra-hepáticos (doenças hepáticas).
Existem várias condições de hiperbilirrubinemia conjugada em crianças :
- Infecções: Virais (hepatite A-E, herpes, adenovírus, citomegalovírus, HIV); bacterianas (sepse, infecções do trato urinário), rubéola, tuberculose, etc.
- Doenças que causam obstrução do canal biliar como atresia biliar, perfuração do canal biliar, colelitíase, etc
- Doenças do sistema endócrino: hipotiroidismo, displasia septoóptica, etc.
- Genética/metabólica: fibrose cística, síndrome de Alagille, alfa-1-antitripsina, etc.
- Desordens de armazenamento: Síndrome de Gaucher, doenças de armazenamento do glicogénio, Nieman-Pick, etc.
- Exposição a muitos medicamentos incluindo ceftriaxona, metotrexato, eritromicina, tetraciclina, etc.
Diagnóstico de Hiperbilirrubinemia Conjugada Pediátrica
História
Para fazer um diagnóstico de hiperbilirrubinemia em crianças, é muito importante fazer um bom histórico clínico, incluindo os pais e os antecedentes patológicos de parentes próximos, a possível existência de consanguinidade entre os pais, possíveis complicações durante a gravidez e o parto, possível exposição a uma doença infecciosa antes ou depois do nascimento (como hepatite, malária, citomegalovírus e leptospirose), entre outras.
Image: “Recém-nascido submetido a fototerapia para tratar a icterícia neonatal” de Martin Pot. Licença: CC BY 3.0
Exame
Exame físico é muito importante no diagnóstico de hiperbilirrubinemia. O sinal principal é icterícia ou descoloração amarelada da esclera, mucosas e da pele. A urina do bebé torna-se escura e, dependendo da causa, as fezes podem ser brancas.
É também extremamente importante realizar um exame neurológico do bebé (reflexos e resposta a estímulos externos) para determinar a aflição do sistema nervoso central (que é conhecida como encefalopatia de kernicterus ou bilirrubina). Isto indica a presença de hiperbilirrubinemia grave.
Outros parâmetros importantes que devem ser examinados incluem parâmetros de crescimento, sinais vitais, sopro cardíaco e respiratório, exame abdominal (a fim de determinar o crescimento anormal do baço ou fígado).
Testes laboratoriais de hiperbilirrubinemia conjugada pediátrica
Os testes laboratoriais necessários são:
- Contagem completa de células sanguíneas, necessária para o rastreamento de hemólise.
- Níveis de aminotransferases do soro são feitos para estudar a função do fígado. Eles incluem aspartato aminotransferase e alanina aminotransferase .
- Rastreio serológico para vírus.
- Fosfatase alcalina, uma vez que um aumento na mesma pode indicar obstrução dos canais biliares.
- Gama-glutamil transpeptidase cujos níveis podem ajudar a diferenciar uma fonte hepática da ALP elevada de outras causas.
- Bilirrubina fracionada.
Em outros termos, testes de função hepática (LFT ou LF) são necessários para o diagnóstico e incluem o tempo de protrombina (PT/INR), tempo de tromboplastina parcial ativada (aPTT), albumina, bilirrubina (direta e indireta) e transaminases hepáticas (AST e ALT). Biópsia e exames de imagem são geralmente reservados para aqueles casos com diagnóstico pouco claro ou descartar patologias obstrutivas.
Causas da hiperbilirrubinemia conjugada pediátrica
Atresia biliar
Atresia biliar é outra causa muito frequente de hiperbilirrubinemia conjugada. O paciente pode ter uma obstrução total ou parcial da árvore biliar extra-hepática em diferentes pontos; no entanto, as partes mais frequentemente afectadas são o ducto hepático e o ducto biliar comum. A obstrução parcial é geralmente causada por uma fibro-inflamação ao longo do ducto biliar.
Esta fibro-inflamação é devida a uma doença perinatal, geralmente uma infecção viral que afecta a mucosa do ducto biliar. Quando a infecção progride, o sistema imunológico responde e há um espessamento epitelial na mucosa que leva à obstrução do ducto. À medida que o tempo avança, a mucosa proliferante desenvolve esclerose e fibrose, causando atresia total do ducto durante o primeiro mês de vida e suas complicações subsequentes.
No diagnóstico da atresia biliar, é também muito importante fazer uma história clínica completa, realizar bons exames físicos e laboratoriais como mencionado anteriormente; no entanto, neste caso, técnicas de imagem (como ultra-som, exame hepatobiliar (HIDA) ou mesmo um colangiograma intra-operatório) podem ser muito úteis no diagnóstico da doença.
Nenhum tratamento médico primário é eficaz na correção da atresia biliar; portanto, uma vez confirmado o diagnóstico, a intervenção cirúrgica é necessária e, em muitos casos, a própria cirurgia pode ser diagnóstica. Ela pode ser feita como um colangiograma intra-operatório ou como uma portoenterostomia de Kasai. Os transplantes hepáticos são reservados para casos graves.
Cistos Coledochal
Finalmente, os cistos coledochal, mais conhecidos como dilatação do ducto biliar congénito, também podem causar hiperbilirrubinemia conjugada. A anomalia congénita mais comum dos cistos coledocolequeais é a dilatação do ducto biliar extra-hepático (1 de cada 100.000-150.000 recém-nascidos). Os sintomas dos cistos coledocolequeais podem apresentar-se em qualquer idade; no entanto, são caracteristicamente observados com icterícia obstrutiva juntamente com dor abdominal em lactentes e crianças. Têm uma predilecção feminina com uma proporção feminina para masculino de cerca de 3-4:1. São mais comuns em certas raças asiáticas.
Tipos de quistos coledocais: Inicialmente, foram descritos três tipos de cistos coledocais, mas Todani e seus associados os classificaram em cinco tipos que são descritos abaixo:
- Tipo I: Existe uma dilatação do canal biliar comum. Pode ser cístico, focal ou fusiforme (visto em 90-95% dos casos).
- Tipo II: Aqui é visto um divertículo do ducto biliar extra-hepático.
- Tipo III: Caracterizado por coledodoceles.
- Tipo IV: É de dois tipos, Tipo IV-A e Tipo IV-B. O Tipo IV-A é o segundo tipo mais comum e é definido como dilatação intra-hepática e extra-hepática da árvore biliar. O Tipo IV-B envolve a malformação rara de múltiplos cistos extra-hepáticos.
- Tipo V: Este tipo também é conhecido como doença de Caroli quando está associado com fibrose hepática. Tipo V inclui cistos intra-hepáticos simples ou múltiplos.
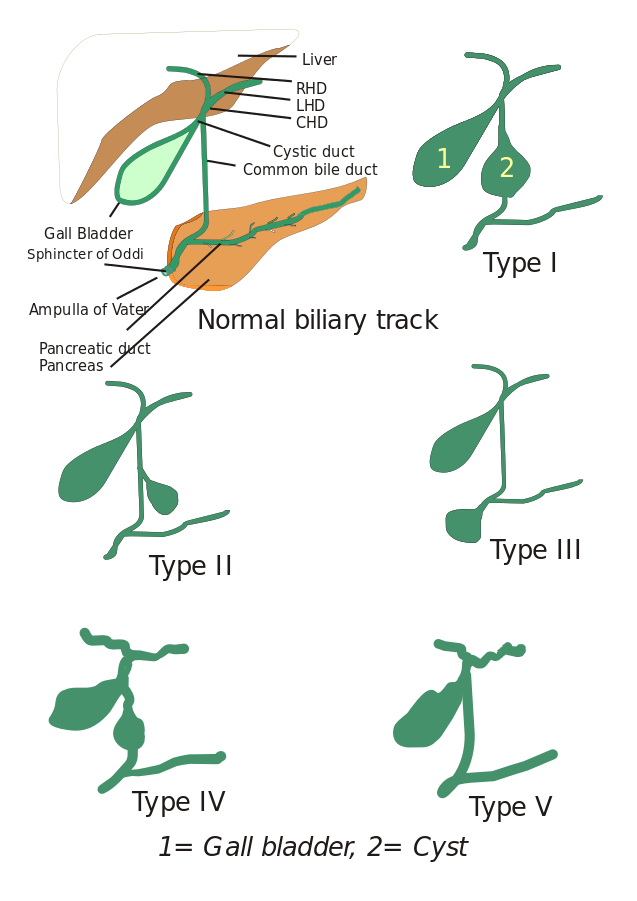
Image: “5 tipos de quistos Choledochal”. Tipo I: Dilatação de ducto biliar extra-hepático; Tipo II: Cisto de ducto biliar comum (CDB); Tipo III: Choledochocele ou dilatação da parte distal da CDB; Tipo IV: Dilatação de ducto extra-hepático e intra-hepático; Tipo V: Doença de Caroli, Dilatação apenas do ducto intra-hepático. CHD: Ducto hepático comum, DLH: Ducto hepático esquerdo e DRH: Ducto hepático direito”. Por mim, Drriad. Licença: CC BY-SA 3.0
É comum em bebés de 1 a 3 meses. Clinicamente, eles apresentam febre, dor no quadrante superior direito do abdômen, fezes dolorosas ou pálidas e hepatomegalia e este quadro é semelhante à atresia biliar. No caso do diagnóstico pré-natal de cisto coledochal, a icterícia só é aparente 1 a 3 semanas após o nascimento. Quando o cisto coledochal se apresenta mais tarde, só se torna clinicamente evidente após 2 anos de idade e pode apresentar uma tríade clássica de dor abdominal, massa abdominal palpável e icterícia; das quais duas são encontradas em quase 85% das crianças no momento da apresentação. Outras características comuns são colangite, pancreatite e peritonite biliar devido à ruptura do cisto.
As investigações laboratoriais, assim como as imagens, são necessárias para um diagnóstico definitivo. As investigações laboratoriais incluem os testes de funções hepáticas convencionais, hemograma e perfis de coagulação. Os estudos de imagem incluem ultra-sonografia, tomografia computadorizada, ressonância magnética, colangiopancreatografia de ressonância magnética e colangiopancreatografia retrógrada endoscópica, entre outros.
Ultrasonografia auxilia no diagnóstico de cistos coledocais e tem uma especificidade muito alta de 97% em crianças, mas uma baixa acurácia diagnóstica antenatal; assim, é usada como modalidade de diagnóstico por imagem primária para icterícia neonatal que persiste mais de 2 semanas pós-parto. Também é útil na diferenciação entre os cistos coledocolecais e a atresia biliar.
O uso de tomografias computadorizadas para o diagnóstico de cistos coledolecais é controverso. Quistos de pequeno tamanho ou coledocele não são detectados nas tomografias, mas são facilmente vistos na colangiopancreatografia por ressonância magnética (MRCP). A tomografia computadorizada pode ser melhor do que a RMC no pós-operatório para detectar a localização da anastomose biliar-entérica e para definir qualquer estenose da mesma.
A colangiopancreatografia por ressonância magnética (RMC) é superior na detecção e definição das lesões. Atualmente, é considerada como o padrão ouro na imagem dos cistos coledocais, porém, tem o inconveniente de não ser capaz de detectar com precisão qualquer unificação pancreático-mobiliar anômala. No entanto, isto não tem um papel importante na determinação do manejo do paciente. Além disso, a sensibilidade da CPRE é maior em adultos do que em lactentes.
A colangiopancreatografia retrógrada endoscópica (CPRE) é a melhor ferramenta para conhecer a anatomia biliar e, portanto, é útil no diagnóstico dos cistos coledocianos.
O tratamento inclui principalmente a intervenção cirúrgica. Um procedimento de drenagem interna conhecido como cistoenterostomia (que poderia ser cistoduodenectomia ou cistojejunostomia) foi feito no passado para o tratamento cirúrgico dos cistos coledociecais; entretanto, várias complicações foram observadas no pós-operatório, como malignidade no cisto remanescente, pancreatite e colangite; assim, elas foram abandonadas.
O procedimento cirúrgico recomendado atualmente é a excisão do cisto e depois a hepáticojejunostomia de Roux-en-Y ou colledochojejunostomia. Devido à excisão do cisto, há uma redução na incidência de formação de estrictura no pós-operatório. Outras alternativas, ou seja, a duodenotomia hepática, foram sugeridas para que a anastomose seja acessível à CPRE em caso de complicações pós-operatórias. Entretanto, o uso da duodenotomia hepática não tem sido amplamente aceito, pois pode causar refluxo biliar e colangite.
Pós excisão de cisto, sondagem e lavagem copiosa dos ductos intra-hepáticos com soro fisiológico é necessário para remover completamente o lodo e possíveis pedras do sistema ductal. Além disso, a obstrução pode estar presente no sistema biliar proximal que pode ser dilatado. Consequentemente, antes da excisão do cisto, uma colangiografia intra-operatória é obrigatória.
Defeitos Genéticos da Hiperbilirrubinemia Conjugada Pediátrica
Existem muitas síndromes hereditárias relacionadas à hiperbilirrubinemia e à colestase intra-hepática. Muitas delas estão relacionadas a mutações genéticas, incluindo o gene SERPINAI (alfa 1- antitripsina), JAG1 (causador da síndrome de Alagille), ATP8B1 (também conhecido como FIC1), ABCB11 (bomba de exportação de sal da bílis), MDR3 (ABCB4), e MRP2 (causador da síndrome de Dubin-Johnson).
1) Síndrome de Alagille
Síndrome de Alagille é um distúrbio genético autossômico dominante que pode afetar muitas partes do corpo. A mutação ocorre no braço curto do cromossomo 20 (20p12). Uma em cada 20 ou 30 mutações ocorre de novo. Um dos órgãos mais afetados na síndrome de Alagille é o fígado e os dutos biliares. Estas malformações dos canais biliares causam acúmulo de bílis no fígado, ou seja, colestase, que causa cicatrizes no fígado, alterando seu funcionamento.
Os sinais e sintomas decorrentes de dano hepático na síndrome de Alagille podem incluir icterícia, comichão na pele e xantomas. No entanto, o coração também é afetado e o paciente pode ter estenose pulmonar. Na estenose pulmonar, há comprometimento do fluxo sanguíneo do coração para os pulmões. Também pode haver doenças cardíacas congênitas coexistentes, como defeitos do septo ventricular, tetralogia de Fallot, etc. O cérebro, medula espinhal, rins e vasos sanguíneos também podem ser afetados.
Síndrome de Alagille tem características faciais. O paciente geralmente tem uma face triangular, uma testa larga e proeminente, ponte nasal larga, olhos profundos e um queixo pequeno e pontiagudo. O diagnóstico é difícil e requer pelo menos 3 das características físicas típicas, a evidência de obstrução do canal biliar (colestase) e uma biópsia hepática.
O tratamento depende da gravidade da doença na síndrome de Alagille. No caso de doença leve, o ácido ursodeoxicólico é administrado para facilitar o fluxo biliar e são administrados anti-histamínicos como a difenidramina para controlar o prurido. Em casos graves, pode ser necessário um transplante de fígado. A suplementação vitamínica foi considerada útil, especialmente com vitaminas A, D, E e K. A suplementação de zinco também é benéfica. Estes pacientes têm uma incapacidade de absorver estas vitaminas e, portanto, a suplementação ajuda a facilitar o crescimento óptimo do paciente.
2) Síndrome de Dubin-Johnson
Esta síndrome é caracterizada por hiperbilirrubinemia crónica conjugada isolada, sem evidência de hemólise devido a um defeito na eliminação da bilirrubinemia conjugada na bílis (colestase intra-hepática). É causada por uma mutação no gene MRP2. É uma condição benigna que não requer tratamento específico.
Síndrome Motora
É uma doença rara caracterizada por hiperbilirrubinemia crônica conjugada e não conjugada sem hemólise. A herança genética ainda não é conhecida. Ocorre devido a um defeito de armazenamento da bilirrubinemia conjugada no fígado, que vaza para o plasma causando hiperbilirrubinemia.
Estudo para escola de medicina e conselhos com Lecturio.
- USMLE Etapa 1
- USMLE Etapa 2
- COMLEX Nível 1
- COMLEX Nível 2
- ENARM
- NEET