
Afbeelding: “Baby op de neonatale intensive care.” door Jacoplane (nou ja, zijn ouders toch) – Eigen werk. Licentie: CC BY-SA 3.0
- Inleiding
- Etiologie van Pediatrische Geconjugeerde Hyperbilirubinemie
- Diagnose van Pediatrische Geconjugeerde Hyperbilirubinemie
- Geschiedenis
- Onderzoek
- Laboratoriumonderzoeken van pediatrische geconjugeerde hyperbilirubinemie
- Cholestatische oorzaken van pediatrische geconjugeerde hyperbilirubinemie
- Biliaire atresie
- Choledochale Cysten
- Genetische Defecten van Pediatrische Geconjugeerde Hyperbilirubinemie
- 1) Alagille-syndroom
- 2) Dubin-Johnson Syndroom
- Rotor Syndrome
Inleiding
Bilirubine is een pigment (tetrapyrrool) dat in de normale katabole route wordt gevormd uit de heemgroep. Het is dus een afvalprodukt van de destructie van rijpe rode bloedcellen. Naargelang van zijn metabolisme kan het worden ingedeeld in geconjugeerde (gecorreleerd met directe bilirubine) en niet-geconjugeerde bilirubine (gecorreleerd met indirecte bilirubine).
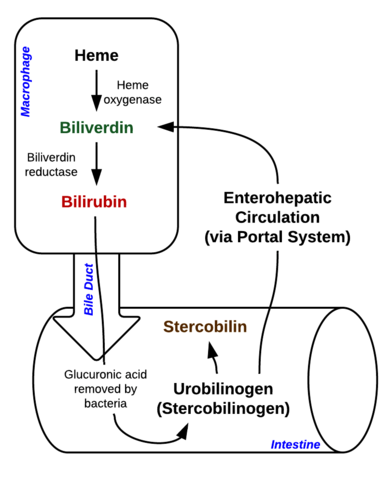
Afbeelding: “Afbraak van Heme in macrofagen en darm” door Johndheathcote. Licentie: CC BY-SA 3.0
Hyperbilirubinemie kan worden gedefinieerd als een verhoogde concentratie bilirubine in het bloed, die tot uiting komt in de vorm van geelzucht (ook wel icterus genoemd). Geelzucht ontstaat wanneer de gele bilirubine pigmenten zich afzetten in de huid, sclera, slijmvliezen en andere weefsels wat leidt tot gelige verkleuring.
Het normale bereik van bilirubine is:
- Serum totaal bilirubine tussen 0,2 en 1,2 mg/dL.
- Serum direct bilirubine tussen 0.1 en 0,4 mg/dL.
Het wordt beschouwd als hyperbilirubinemie wanneer:
- Serum directe bilirubine ≥ 1 mg/dL is (als totale bilirubine < 5mg/dL is).
- Serum geconjugeerde bilirubine > 20% van totale bilirubine is (als totale bilirubine > 5mg/dL is).
Etiologie van Pediatrische Geconjugeerde Hyperbilirubinemie
Bij sommige pasgeborenen, vooral te vroeg geboren baby’s, kan een lichte toename van bilirubine als normaal worden beschouwd; dit moet echter altijd worden onderzocht omdat een hoog bilirubinegehalte ten koste van geconjugeerd bilirubine nooit als normaal kan worden beschouwd.
Er zijn vele ziekten die hyperbilirubinemie kunnen veroorzaken, maar ze kunnen grofweg in twee groepen worden ingedeeld: extrahepatische aandoeningen (prehepatische oorzaken zoals hemolyse door intrinsieke defecten van rode bloedcellen of extrinsieke oorzaken die leiden tot ruptuur van rode bloedcellen, of posthepatische oorzaken waaronder obstructie van de galwegen) en intrahepatische aandoeningen (leverziekten).
Er zijn verschillende aandoeningen van geconjugeerde hyperbilirubinemie bij kinderen :
- Infecties: Virale (hepatitis A-E, herpes, adenovirus, cytomegalovirus, HIV); bacteriële (sepsis, infecties van de urinewegen), rubella, tuberculose, enz.
- Aandoeningen die obstructie van de galwegen veroorzaken, zoals atresie van de galwegen, perforatie van de galwegen, cholelithiasis, enz
- Aandoeningen van het endocriene systeem: hypothyreoïdie, septooptische dysplasie, enz.
- Genetisch/metabolisch: mucoviscidose, syndroom van Alagille, alfa-1-antitrypsine, enz.
- Storingsstoornissen: Gaucher syndroom, glycogeen opslag ziekten, Nieman-Pick, enz.
- Blootstelling aan veel medicijnen, waaronder ceftriaxon, methotrexaat, erytromycine, tetracycline, enz.
Diagnose van Pediatrische Geconjugeerde Hyperbilirubinemie
Geschiedenis
Om de diagnose hyperbilirubinemie bij kinderen te stellen is het van groot belang een goede klinische anamnese af te nemen, inclusief de pathologische antecedenten van de ouders en naaste familieleden, het mogelijke bestaan van consanguiniteit tussen de ouders, mogelijke complicaties tijdens de zwangerschap en de bevalling, mogelijke blootstelling aan een infectieziekte voor of na de geboorte (zoals onder andere hepatitis, malaria, cytomegalovirus en leptospirose).
Afbeelding: “Pasgeborene die fototherapie ondergaat om neonatale geelzucht te behandelen” door Martin Pot. Licentie: CC BY 3.0
Onderzoek
Lichamelijk onderzoek is zeer belangrijk bij de diagnose van hyperbilirubinemie. Het belangrijkste teken is geelzucht of gelige verkleuring van de sclera, slijmvliezen en de huid. De urine van de baby wordt donker en, afhankelijk van de oorzaak, kan de ontlasting wit zijn.
Het is ook uiterst belangrijk om een neurologisch onderzoek van de baby uit te voeren (reflexen en reactie op externe prikkels) om de aandoening van het centrale zenuwstelsel vast te stellen (dat bekend staat als kernicterus of bilirubine encefalopathie). Dit wijst op de aanwezigheid van ernstige hyperbilirubinemie.
Andere belangrijke parameters die moeten worden onderzocht zijn groeiparameters, vitale functies, hart- en ademhalingsgeruis, abdominaal onderzoek (om de abnormale groei van de milt of de lever vast te stellen).
Laboratoriumonderzoeken van pediatrische geconjugeerde hyperbilirubinemie
De vereiste laboratoriumonderzoeken zijn:
- Complete bloedceltelling die nodig is voor de screening van hemolyse.
- Serum aminotransferasen niveaus worden gedaan om de functie van de lever te bestuderen. Zij omvatten aspartaataminotransferase en alanineaminotransferase.
- Serologisch onderzoek naar virussen.
- Alkalische fosfatase, aangezien een stijging ervan kan wijzen op een obstructie van de galwegen.
- Gamma-glutamyltranspeptidase, waarvan de niveaus kunnen helpen om een leverbron van het verhoogde ALP van andere oorzaken te onderscheiden.
- gefractioneerde bilirubine.
In andere termen zijn leverfunctietests (LFT’s of LF’s) noodzakelijk voor de diagnose en deze omvatten protrombinetijd (PT/INR), geactiveerde gedeeltelijke tromboplastinetijd (aPTT), albumine, bilirubine (direct en indirect), en levertransaminasen (AST en ALT). Biopsie en beeldvormend onderzoek worden gewoonlijk gereserveerd voor gevallen met een onduidelijke diagnose of om obstructieve pathologieën te discuseren.
Cholestatische oorzaken van pediatrische geconjugeerde hyperbilirubinemie
Biliaire atresie
Biliaire atresie is een andere zeer frequente oorzaak van geconjugeerde hyperbilirubinemie. De patiënt kan een volledige of gedeeltelijke obstructie van de extrahepatische galboom hebben op verschillende punten; de meest voorkomende delen die zijn aangetast zijn echter de leverbuis en de gewone galbuis. Gedeeltelijke obstructie wordt gewoonlijk veroorzaakt door een fibro-inflammatie langs de galbuis.
Deze fibro-inflammatie is het gevolg van een perinatale ziekte, meestal een virale infectie die het slijmvlies van de galbuis aantast. Wanneer de infectie voortschrijdt, reageert het immunologische systeem en ontstaat er een epitheliale verdikking in de mucosa die leidt tot obstructie van het kanaal. Naarmate de tijd vordert, ontwikkelt de woekerende mucosa sclerose en fibrose, waardoor totale kanaalatresie ontstaat tijdens de eerste levensmaand en de daaropvolgende complicaties.
Bij de diagnose van biliaire atresie is het ook zeer belangrijk om een volledige klinische anamnese af te nemen, goed lichamelijk onderzoek te verrichten en laboratoriumonderzoek te doen, zoals eerder vermeld; in dit geval kunnen beeldvormende technieken (zoals echografie, hepatobiliary scan (HIDA) of zelfs een intraoperatief cholangiogram) echter zeer nuttig zijn bij de diagnose van de ziekte.
Geen enkele primaire medische behandeling is doeltreffend voor de correctie van biliaire atresie; daarom is, zodra de diagnose is bevestigd, een chirurgische ingreep noodzakelijk en in veel gevallen kan de operatie zelf diagnostisch zijn. Dit kan gebeuren in de vorm van een intraoperatief cholangiogram of een Kasai portoenterostomie. Levertransplantaties zijn voorbehouden voor ernstige gevallen.
Choledochale Cysten
Ten slotte kunnen ook choledochale cysten, beter bekend als congenitale galwegdilatatie, geconjugeerde hyperbilirubinemie veroorzaken. De meest voorkomende aangeboren afwijking van choledochale cysten is de vergroting van de extrahepatische galbuis (1 op elke 100.000-150.000 pasgeborenen). De symptomen van choledochale cysten kunnen zich op elke leeftijd voordoen; ze worden echter meestal gezien met obstructieve geelzucht samen met abdominale pijn bij zuigelingen en kinderen. Ze hebben een vrouwelijke voorkeur met een vrouw/man verhouding van ongeveer 3-4:1. Ze komen meer voor bij bepaalde Aziatische rassen.
Types van choledochale cysten: Aanvankelijk werden drie typen choledochale cysten beschreven, maar Todani en zijn medewerkers hebben deze verder ingedeeld in vijf typen, die hieronder worden beschreven:
- Type I: Er is verwijding van het gemeenschappelijk galkanaal. Deze kan cystisch, focaal of fusiform zijn (gezien in 90-95% van de gevallen).
- Type II: Hier wordt een diverticulum van de extrahepatische galbuis gezien.
- Type III: Gekenmerkt door choledochoceles.
- Types IV: Er zijn twee typen, Type IV-A en Type IV-B. Type IV-A is het op één na meest voorkomende type en wordt gedefinieerd als zowel intrahepatische als extrahepatische verwijding van de galwegen. Type IV-B betreft de zeldzame misvorming van meerdere extrahepatische cysten.
- Type V: Dit type staat ook bekend als de ziekte van Caroli wanneer het gepaard gaat met leverfibrose. Type V omvat enkele of meerdere intrahepatische cysten.
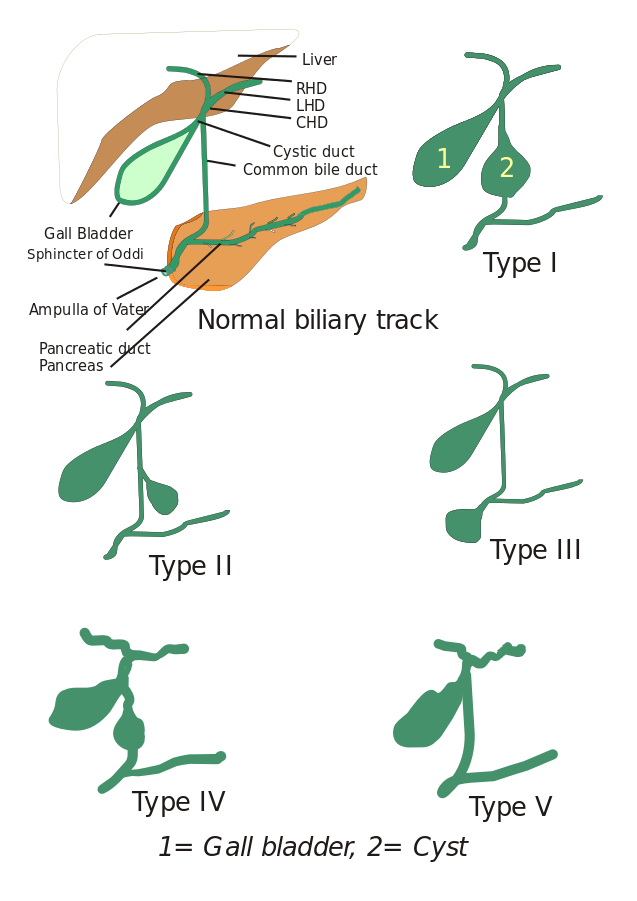
Afbeelding: “5 types van Choledochale cysten. Type I: Dilatatie van extrahepatische galbuis; Type II: Cyste van gemeenschappelijke galbuis (CBD); Type III: Choledochocele of dilatatie van distale deel van CBD, type IV: Dilatatie van zowel extrahepatische als intrahepatische galbuis; Type V: Ziekte van Caroli, Dilatatie van alleen de intrahepatische ductus. CHD: Gewone leverbuis, LHD: Linker leverkanaal en RHD: Rechter hepatische duct.” Door I, Drriad. Licentie: CC BY-SA 3.0
Het wordt vaak gezien bij zuigelingen van 1 tot 3 maanden. Klinisch presenteren zij zich met koorts, pijn in het rechter bovenkwadrant van de buik, acholische of bleke ontlasting en hepatomegalie en dit beeld is vergelijkbaar met biliaire atresie. In het geval van prenatale diagnose van choledochale cyste, is geelzucht niet zichtbaar tot 1 tot 3 weken na de geboorte. Wanneer de choledochale cyste zich later voordoet, wordt deze pas na de leeftijd van 2 jaar klinisch duidelijk en kan deze zich presenteren met een klassiek trias van buikpijn, palpabele buikmassa en geelzucht, waarvan er twee worden aangetroffen bij bijna 85% van de kinderen op het moment van presentatie. Andere veel voorkomende kenmerken zijn cholangitis, pancreatitis en biliaire peritonitis als gevolg van ruptuur van de cyste.
Laboratoriumonderzoeken, evenals beeldvorming, zijn noodzakelijk voor een definitieve diagnose. Laboratoriumonderzoeken omvatten de conventionele leverfunctietests, CBC en stollingsprofielen. Beeldvormend onderzoek omvat onder meer echografie, computertomografie, magnetische resonantiebeeldvorming, magnetische resonantiecholangiopancreatografie en endoscopische retrograde cholangiopancreatografie.
Echografie helpt bij de diagnose van choledochale cysten en heeft een zeer hoge specificiteit van 97% bij kinderen, maar een lage diagnostische nauwkeurigheid antenataal; daarom wordt het gebruikt als een primaire diagnostische beeldvormingsmodaliteit voor neonatale geelzucht die langer aanhoudt dan 2 weken na de geboorte. Het is ook nuttig bij de differentiatie tussen choledochale cysten en biliaire atresie.
Het gebruik van computertomografische scans voor de diagnose van choledochale cysten is controversieel. Kleine cysten of choledochocele worden op CT-scans gemist, maar zijn goed zichtbaar op magnetische resonantie cholangiopancreatografie (MRCP). CT scanning kan beter zijn dan MRCP in de postoperatieve periode voor het opsporen van de plaats van de biliary-enteric anastomosis en voor het definiëren van eventuele stenose daarvan.
Magnetische resonantie cholangiopancreatografie (MRCP) is superieur in het opsporen en definiëren van laesies. Tegenwoordig wordt MRCP beschouwd als de gouden standaard voor de beeldvorming van choledochale cysten; het nadeel is echter dat hiermee een afwijkende pancreas-biliaire unificatie niet nauwkeurig kan worden gedetecteerd. Dit speelt echter geen grote rol bij het bepalen van de behandeling van de patiënt. Bovendien is de gevoeligheid van MRCP groter bij volwassenen dan bij zuigelingen.
Endoscopische retrograde cholangiopancreatografie (ERCP) is het beste instrument om de anatomie van de galwegen te kennen en is dus nuttig bij de diagnose van choledochale cysten.
De behandeling omvat in de eerste plaats een chirurgische ingreep. Een interne drainage procedure bekend als een cyste-enterostomie (die ofwel cyste-duodenectomie of cyste-jejunostomie kon zijn) werd in het verleden gedaan voor chirurgische behandeling van choledochale cysten; er werden echter verschillende complicaties postoperatief gezien, zoals kwaadaardigheid in de resterende cyste, pancreatitis, en cholangitis; daarom werden ze verlaten.
Op dit moment is de aanbevolen chirurgische procedure cyste-excisie en vervolgens Roux-en-Y hepaticojejunostomie of choledochojejunostomie. Door de cyste-excisie is er een vermindering van de incidentie van strictuurvorming postoperatief. Andere alternatieven, zoals leverduodenotomie, zijn voorgesteld zodat de anastomose toegankelijk is voor ERCP in geval van postoperatieve complicaties. Het gebruik van duodenotomie van de lever is echter niet algemeen aanvaard omdat het biliaire reflux en cholangitis kan veroorzaken.
Na de cyste-excisie is sonderen en overvloedig spoelen van de intrahepatische kanalen met zoutoplossing noodzakelijk om slib en mogelijke stenen volledig uit het ductale systeem te verwijderen. Bovendien kan de obstructie aanwezig zijn in het proximale biliaire systeem dat kan worden verwijd. Bijgevolg is vóór de excisie van de cyste een intraoperatieve cholangiografie verplicht.
Genetische Defecten van Pediatrische Geconjugeerde Hyperbilirubinemie
Er zijn heel wat erfelijke syndromen die verband houden met hyperbilirubinemie en intrahepatische cholestase. Velen van hen zijn gerelateerd aan genetische mutaties, waaronder het gen SERPINAI (alfa 1-antitrypsine), JAG1 (dat het syndroom van Alagille veroorzaakt), ATP8B1 (ook bekend als FIC1), ABCB11 (exportpomp voor galzouten), MDR3 (ABCB4) en MRP2 (dat het syndroom van Dubin-Johnson veroorzaakt).
1) Alagille-syndroom
Alagille-syndroom is een autosomaal dominante genetische aandoening die vele delen van het lichaam kan aantasten. De mutatie treedt op in de korte arm van chromosoom 20 (20p12). Eén op elke 20 of 30 mutaties treedt de novo op. Een van de meest aangetaste organen bij het syndroom van Alagille is de lever en de galwegen. Deze misvormingen van de galwegen veroorzaken ophoping van gal in de lever (cholestase), waardoor littekenvorming in de lever optreedt en de lever niet meer goed functioneert.
De tekenen en symptomen die het gevolg zijn van leverbeschadiging bij het syndroom van Alagille kunnen geelzucht, jeukende huid en xanthomen zijn. Het hart wordt echter ook aangetast en de patiënt kan pulmonale stenose hebben. Bij pulmonale stenose is er een verminderde bloedstroom van het hart naar de longen. Er kunnen ook aangeboren hartziekten zijn, zoals ventrikelseptumdefecten, tetralogie van Fallot, enz. Ook de hersenen, het ruggenmerg, de nieren en de bloedvaten kunnen zijn aangedaan.
Alagillesyndroom heeft karakteristieke gelaatstrekken. De patiënt heeft meestal een driehoekig gezicht, een breed en prominent voorhoofd, een brede neusbrug, diepliggende ogen, en een kleine, spitse kin. De diagnose is moeilijk en vereist ten minste 3 van de typische lichamelijke kenmerken, het bewijs van galwegobstructie (cholestase) en een leverbiopsie.
De behandeling is afhankelijk van de ernst van de ziekte bij het syndroom van Alagille. Bij lichte ziekte wordt ursodeoxycholzuur toegediend om de biliaire doorstroming te bevorderen en antihistaminica zoals diphenhydramine om de pruritus onder controle te houden. In ernstige gevallen kan een levertransplantatie nodig zijn. Vitaminesupplementen blijken nuttig te zijn, vooral vitamine A, D, E en K. Ook zinksupplementen zijn nuttig. Deze patiënten zijn niet in staat deze vitamines op te nemen en dus helpt suppletie bij het vergemakkelijken van een optimale groei van de patiënt.
2) Dubin-Johnson Syndroom
Dit syndroom wordt gekenmerkt door geïsoleerde geconjugeerde chronische hyperbilirubinemie, zonder aanwijzingen voor hemolyse als gevolg van een defect in de eliminatie van geconjugeerde bilirubine in de gal (intrahepatische cholestase). Het wordt veroorzaakt door een mutatie in het MRP2-gen. Het is een goedaardige aandoening die geen specifieke behandeling vereist.
Rotor Syndrome
Het is een zeldzame aandoening die wordt gekenmerkt door chronische geconjugeerde en niet-geconjugeerde hyperbilirubinemie zonder hemolyse. De genetische overerving is nog niet bekend. Het treedt op als gevolg van een defect in de opslag van geconjugeerde bilirubine in de lever, die lekt in het plasma waardoor hyperbilirubinemie ontstaat.
Studie voor medische school en besturen met Lecturio.
- USMLE Stap 1
- USMLE Stap 2
- COMLEX Level 1
- COMLEX Level 2
- ENARM
- NEET