
Immagine: “Bambino nel reparto di terapia intensiva neonatale” di Jacoplane (beh, i suoi genitori comunque) – Opera propria. Licenza: CC BY-SA 3.0
- Introduzione
- Eziologia dell’iperbilirubinemia coniugata pediatrica
- Diagnosi dell’iperbilirubinemia coniugata pediatrica
- Storia
- Esame
- Test di laboratorio dell’iperbilirubinemia coniugata pediatrica
- Cause colestatiche di iperbilitubinemia coniugata pediatrica
- Atresia biliare
- Cisti coledochali
- Difetti genetici dell’iperbilirubinemia coniugata pediatrica
- 1) Sindrome di Alagille
- 2) Sindrome di Dubin-Johnson
- Sindrome di Rotor
Introduzione
La bilirubina è un pigmento (tetrapirrolo) che si forma nella normale via catabolica dal gruppo eme. È quindi un prodotto di scarto della distruzione dei globuli rossi maturi. Secondo il suo metabolismo, può essere classificata come bilirubina coniugata (correlata alla bilirubina diretta) e non coniugata (correlata alla bilirubina indiretta).
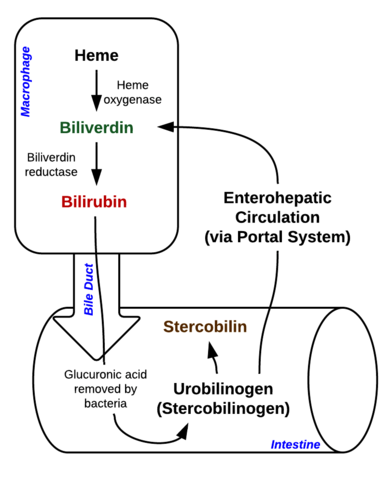
Immagine: “Scomposizione dell’eme nei macrofagi e nell’intestino” di Johndheathcote. Licenza: CC BY-SA 3.0
L’iperbilirubinemia può essere definita come una concentrazione elevata di bilirubina nel sangue, che si esprime sotto forma di ittero (chiamato anche ittero). L’ittero appare quando i pigmenti gialli della bilirubina si depositano nella pelle, nella sclera, nelle membrane mucose e in altri tessuti, portando a una decolorazione giallastra.
Il range normale della bilirubina è:
- Bilirubina totale del siero tra 0,2 e 1,2 mg/dL.
- Bilirubina diretta del siero tra 0.1 e 0,4 mg/dL.
Si considera iperbilirubinemia quando:
- La bilirubina diretta del siero è ≥ 1 mg/dL (se la bilirubina totale è < 5mg/dL).
- La bilirubina coniugata del siero è > 20% della bilirubina totale (se la bilirubina totale è > 5mg/dL).
Eziologia dell’iperbilirubinemia coniugata pediatrica
In alcuni neonati, specialmente i prematuri, un leggero aumento della bilirubina può essere considerato normale; tuttavia, deve sempre essere studiato perché un alto livello di bilirubina a scapito della bilirubina coniugata non può mai essere considerato normale.
Ci sono molte malattie che possono causare iperbilirubinemia, ma possono essere ampiamente classificate in due gruppi: disturbi extraepatici (cause pre-epatiche come l’emolisi dovuta a difetti intrinseci dei globuli rossi o cause estrinseche che portano alla rottura dei globuli rossi, o cause post-epatiche tra cui l’ostruzione del passaggio biliare) e disturbi intraepatici (malattie epatiche).
Ci sono diverse condizioni di iperbilirubinemia coniugata nei bambini :
- Infezioni: Virali (epatite A-E, herpes, adenovirus, citomegalovirus, HIV); batteriche (sepsi, infezioni del tratto urinario), rosolia, tubercolosi, ecc.
- Disordini che causano ostruzione del dotto biliare come atresia biliare, perforazione del dotto biliare, colelitiasi, ecc
- Malattie del sistema endocrino: ipotiroidismo, displasia settica, ecc.
- Genetico/metabolico: fibrosi cistica, sindrome di Alagille, alfa-1-antitripsina, ecc: Sindrome di Gaucher, malattie da accumulo di glicogeno, Nieman-Pick, ecc.
- Esposizione a molti farmaci tra cui ceftriaxone, metotrexato, eritromicina, tetraciclina, ecc.
Diagnosi dell’iperbilirubinemia coniugata pediatrica
Storia
Per fare una diagnosi di iperbilirubinemia nei bambini, è molto importante fare una buona storia clinica, compresi gli antecedenti patologici dei genitori e dei parenti stretti, l’eventuale esistenza di consanguineità tra i genitori, eventuali complicazioni durante la gravidanza e il parto, eventuale esposizione a una malattia infettiva prima o dopo la nascita (come epatite, malaria, citomegalovirus e leptospirosi) tra le altre.
Immagine: “Neonato sottoposto a fototerapia per trattare l’ittero neonatale” di Martin Pot. Licenza: CC BY 3.0
Esame
L’esame fisico è molto importante nella diagnosi di iperbilirubinemia. Il segno principale è l’ittero o lo scolorimento giallastro della sclera, delle membrane mucose e della pelle. L’urina del bambino diventa scura e, a seconda della causa, le feci possono essere bianche.
È anche estremamente importante eseguire un esame neurologico del bambino (riflessi e risposta agli stimoli esterni) per determinare l’afflizione del sistema nervoso centrale (che è conosciuta come kernicterus o encefalopatia da bilirubina). Questo indica la presenza di una grave iperbilirubinemia.
Altri parametri importanti che devono essere esaminati sono i parametri di crescita, i segni vitali, il soffio cardiaco e respiratorio, l’esame addominale (per determinare la crescita anormale della milza o del fegato).
Test di laboratorio dell’iperbilirubinemia coniugata pediatrica
I test di laboratorio richiesti sono:
- Conteggio completo delle cellule del sangue che è necessario per lo screening dell’emolisi.
- I livelli di aminotransferasi nel siero sono fatti per studiare la funzione del fegato. Comprendono l’aspartato aminotransferasi e l’alanina aminotransferasi.
- Schermo sierologico per i virus.
- Fosfatasi alcalina poiché un suo aumento può indicare un’ostruzione dei dotti biliari.
- Gamma-glutamil transpeptidasi i cui livelli possono aiutare a differenziare una fonte epatica di ALP elevata da altre cause.
- Bilirubina frazionata.
In altri termini, i test di funzionalità epatica (LFT o LF) sono necessari per la diagnosi e comprendono tempo di protrombina (PT/INR), tempo di tromboplastina parziale attivato (aPTT), albumina, bilirubina (diretta e indiretta), e transaminasi epatiche (AST e ALT). La biopsia e gli studi di imaging sono di solito riservati ai casi con una diagnosi non chiara o per scartare le patologie ostruttive.
Cause colestatiche di iperbilitubinemia coniugata pediatrica
Atresia biliare
L’atresia biliare è un’altra causa molto frequente di iperbilirubinemia coniugata. Il paziente può avere un’ostruzione completa o parziale dell’albero biliare extraepatico in diversi punti; tuttavia, le parti più comuni interessate sono il dotto epatico e il dotto biliare comune. L’ostruzione parziale è solitamente causata da una fibro-infiammazione lungo il dotto biliare.
Questa fibro-infiammazione è dovuta a una malattia perinatale, solitamente un’infezione virale che colpisce la mucosa del dotto biliare. Quando l’infezione progredisce, il sistema immunologico risponde e c’è un ispessimento epiteliale nella mucosa che porta all’ostruzione del dotto. Con il passare del tempo, la mucosa proliferante sviluppa sclerosi e fibrosi, causando l’atresia totale del dotto nel primo mese di vita e le sue successive complicazioni.
Nella diagnosi di atresia biliare, è anche molto importante prendere una storia clinica completa, eseguire un buon esame fisico e test di laboratorio come menzionato in precedenza; tuttavia, in questo caso, le tecniche di imaging (come ecografia, scansione epatobiliare (HIDA) o anche un colangiogramma intraoperatorio) possono essere molto utili nella diagnosi della malattia.
Nessun trattamento medico primario è efficace nella correzione dell’atresia biliare; quindi, una volta confermata la diagnosi, è necessario un intervento chirurgico e, in molti casi, l’intervento stesso può essere diagnostico. Può essere fatto come un colangiogramma intraoperatorio o una portoenterostomia Kasai. Il trapianto di fegato è riservato ai casi gravi.
Cisti coledochali
Infine, le cisti coledochali, meglio conosciute come dilatazione congenita dei dotti biliari, possono anche causare iperbilirubinemia coniugata. L’anomalia congenita più comune delle cisti coledochali è l’allargamento del dotto biliare extraepatico (1 ogni 100.000-150.000 neonati). I sintomi delle cisti coledocali possono presentarsi a qualsiasi età; tuttavia, sono caratteristici dell’ittero ostruttivo insieme al dolore addominale nei neonati e nei bambini. Hanno una predilezione femminile con un rapporto femmina-maschio di circa 3-4:1. Sono più comuni in alcune razze asiatiche.
Tipi di cisti coledocali: Inizialmente, sono stati descritti tre tipi di cisti coledocali, ma Todani e soci li hanno ulteriormente classificati in cinque tipi che sono descritti di seguito:
- Tipo I: C’è una dilatazione del dotto biliare comune. Potrebbe essere cistico, focale o fusiforme (visto nel 90-95% dei casi).
- Tipo II: Un diverticolo del dotto biliare extraepatico è visto qui.
- Tipo III: Caratterizzato da coledoceli.
- Tipi IV: È di due tipi, Tipo IV-A e Tipo IV-B. Il tipo IV-A è il secondo tipo più comune ed è definito come una dilatazione sia intraepatica che extraepatica dell’albero biliare. Il tipo IV-B comporta la rara malformazione di cisti extraepatiche multiple.
- Tipo V: Questo tipo è anche conosciuto come malattia di Caroli quando è associato a fibrosi epatica. Il tipo V include cisti intraepatiche singole o multiple.
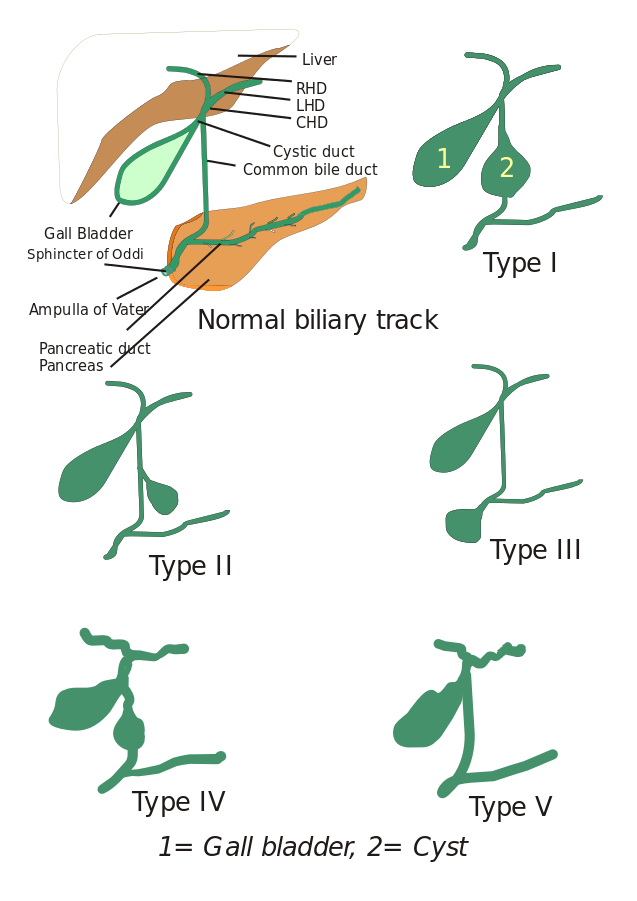
Immagine: “5 tipi di cisti coledocali. Tipo I: Dilatazione del dotto biliare extraepatico; Tipo II: Cisti del dotto biliare comune (CBD); Tipo III: Coledochocele o dilatazione della parte distale del CBD, tipo IV: Dilatazione sia del dotto extraepatico che intraepatico; Tipo V: Malattia di Caroli, Dilatazione del solo dotto intraepatico. CHD: dotto epatico comune, LHD: Dotto epatico sinistro e RHD: Dotto epatico destro”. Da I, Drriad. Licenza: CC BY-SA 3.0
E’ comunemente visto nei bambini da 1 a 3 mesi. Clinicamente, si presentano con febbre, dolore nel quadrante superiore destro dell’addome, feci acoliche o pallide ed epatomegalia e questo quadro è simile all’atresia biliare. In caso di diagnosi prenatale di cisti coledochale, l’ittero non è evidente fino a 1 o 3 settimane dopo la nascita. Quando la cisti coledochale si presenta più tardi, non diventa clinicamente evidente fino a dopo i 2 anni di età e può presentarsi con una classica triade di dolore addominale, massa addominale palpabile e ittero; di cui due sono presenti in quasi l’85% dei bambini al momento della presentazione. Altre caratteristiche comuni sono la colangite, la pancreatite e la peritonite biliare dovuta alla rottura della cisti.
Le indagini di laboratorio, così come l’imaging, sono necessarie per una diagnosi definitiva. Le indagini di laboratorio comprendono i test convenzionali di funzionalità epatica, l’emocromo e i profili di coagulazione. Gli studi di imaging comprendono l’ecografia, la tomografia computerizzata, la risonanza magnetica, la colangiopancreatografia a risonanza magnetica e la colangiopancreatografia retrograda endoscopica tra gli altri.
L’ecografia aiuta nella diagnosi delle cisti coledocali e ha una specificità molto alta del 97% nei bambini, ma una bassa accuratezza diagnostica prenatale; quindi, è usata come modalità di imaging diagnostico primario per l’ittero neonatale che persiste più di 2 settimane dopo la nascita. È anche utile nella differenziazione tra cisti coledochali e atresia biliare.
L’uso della tomografia computerizzata per la diagnosi di cisti coledochali è controverso. Le cisti di piccole dimensioni o il coledocele sono mancati alla TAC ma sono facilmente visibili alla colangiopancreatografia a risonanza magnetica (MRCP). La TAC può essere migliore della MRCP nel periodo postoperatorio per individuare la posizione dell’anastomosi bilio-enterica e per definire eventuali stenosi della stessa.
La colangiopancreatografia a risonanza magnetica (MRCP) è superiore per individuare e definire le lesioni. Al giorno d’oggi, è considerata come il gold standard nell’imaging delle cisti coledocali; tuttavia, ha l’inconveniente di non essere in grado di rilevare con precisione qualsiasi unificazione pancreatico-biliare anomala. Tuttavia, questo non gioca un ruolo importante nel determinare la gestione del paziente. Inoltre, la sensibilità della MRCP è maggiore negli adulti che nei bambini.
La colangiopancreatografia retrograda endoscopica (ERCP) è lo strumento migliore per conoscere l’anatomia biliare ed è quindi utile nella diagnosi delle cisti coledochali.
Il trattamento comprende principalmente l’intervento chirurgico. Una procedura di drenaggio interno nota come una cisti-enterostomia (che potrebbe essere o cisti-duodenectomia o cisti-giunostomia) è stata fatta in passato per il trattamento chirurgico delle cisti coledocali; tuttavia, varie complicazioni sono state viste dopo l’intervento come la malignità nella cisti rimanente, pancreatite e colangite; quindi, sono state abbandonate.
Attualmente, la procedura chirurgica raccomandata è l’escissione della cisti e poi l’epatico-giunostomia Roux-en-Y o la coledo-chojejunostomia. A causa dell’escissione della cisti, c’è una riduzione dell’incidenza della formazione di stenosi post-operatoria. Altre alternative, come la duodenotomia epatica, sono state suggerite in modo che l’anastomosi sia accessibile alla ERCP in caso di complicazioni postoperatorie. Tuttavia, l’uso della duodenotomia epatica non è stato ampiamente accettato in quanto può causare reflusso biliare e colangite.
Dopo l’escissione della cisti, è necessario sondare e lavare abbondantemente i dotti intraepatici con soluzione fisiologica per rimuovere completamente il fango ed eventuali calcoli dal sistema duttale. Inoltre, l’ostruzione può essere presente nel sistema biliare prossimale che può essere dilatato. Di conseguenza, prima dell’escissione della cisti, è obbligatoria una colangiografia intraoperatoria.
Difetti genetici dell’iperbilirubinemia coniugata pediatrica
Ci sono molte sindromi ereditarie legate all’iperbilirubinemia e colestasi intraepatica. Molti di loro sono legati a mutazioni genetiche tra cui il gene SERPINAI (alfa 1- antitripsina), JAG1 (che causa la sindrome di Alagille), ATP8B1 (noto anche come FIC1), ABCB11 (pompa di esportazione dei sali biliari), MDR3 (ABCB4), e MRP2 (che causa la sindrome di Dubin-Johnson).
1) Sindrome di Alagille
La sindrome di Alagille è una malattia genetica autosomica dominante che può colpire molte parti del corpo. La mutazione avviene nel braccio corto del cromosoma 20 (20p12). Una mutazione su 20 o 30 si verifica de novo. Uno degli organi più colpiti nella sindrome di Alagille è il fegato e i dotti biliari. Queste malformazioni dei dotti biliari causano l’accumulo di bile nel fegato, cioè la colestasi, che causa la cicatrizzazione del fegato alterando così il suo funzionamento.
I segni e i sintomi derivanti dal danno epatico nella sindrome di Alagille possono includere ittero, prurito della pelle e xantomi. Tuttavia, anche il cuore è colpito e il paziente può avere una stenosi polmonare. Nella stenosi polmonare, c’è una riduzione del flusso di sangue dal cuore ai polmoni. Ci possono essere anche malattie cardiache congenite coesistenti, come difetti del setto ventricolare, tetralogia di Fallot ecc. Anche il cervello, il midollo spinale, i reni e i vasi sanguigni possono essere colpiti.
La sindrome di Agille ha caratteristiche facciali. Il paziente di solito ha un viso triangolare, una fronte ampia e prominente, un ampio ponte nasale, occhi infossati e un mento piccolo e appuntito. La diagnosi è difficile e richiede almeno 3 delle caratteristiche fisiche tipiche, la prova dell’ostruzione del dotto biliare (colestasi) e una biopsia epatica.
Il trattamento dipende dalla gravità della malattia nella sindrome di Alagille. In caso di malattia lieve, si somministra acido ursodeossicolico per facilitare il flusso biliare e si danno antistaminici come la difenidramina per controllare il prurito. Per i casi gravi, può essere necessario un trapianto di fegato. L’integrazione di vitamine si è rivelata utile, specialmente con le vitamine A, D, E e K. Anche l’integrazione di zinco è utile. Questi pazienti hanno un’incapacità di assorbire queste vitamine e quindi l’integrazione aiuta a facilitare la crescita ottimale del paziente.
2) Sindrome di Dubin-Johnson
Questa sindrome è caratterizzata da iperbilirubinemia cronica coniugata isolata, senza evidenza di emolisi a causa di un difetto nell’eliminazione della bilirubina coniugata nella bile (colestasi intraepatica). È causata da una mutazione nel gene MRP2. È una condizione benigna che non richiede un trattamento specifico.
Sindrome di Rotor
È una malattia rara caratterizzata da iperbilirubinemia cronica coniugata e non coniugata senza emolisi. L’eredità genetica non è ancora conosciuta. Si verifica a causa di un difetto di stoccaggio della bilirubina coniugata nel fegato, che fuoriesce nel plasma causando l’iperbilirubinemia.
Studiare per la scuola medica e gli esami con Lecturio.
- USMLE Step 1
- USMLE Step 2
- COMLEX Livello 1
- COMLEX Livello 2
- ENARM
- NEET