
Obraz: „Baby in the neonatal intensive care unit.” by Jacoplane (well, his parents anyway) – Own work. Licencja: CC BY-SA 3.0
- Wprowadzenie
- Etiologia pediatrycznej hiperbilirubinemii sprzężonej
- Diagnostyka pediatrycznej hiperbilirubinemii sprzężonej
- Historia
- Badanie
- Badania laboratoryjne w pediatrycznej hiperbilirubinemii sprzężonej
- Cholestatyczne przyczyny pediatrycznej sprzężonej hiperbilirubinemii
- Atrezja dróg żółciowych
- Torbiele żółciowe
- Defekty genetyczne dziecięcej hiperbilirubinemii sprzężonej
- 1) Zespół Alagille’a
- 2) Zespół Dubina-Johnsona
- Zespół Rotora
Wprowadzenie
Bilirubina to barwnik (tetrapyrol), który powstaje w normalnym szlaku katabolicznym z grupy hemowej. Jest więc produktem odpadowym niszczenia dojrzałych krwinek czerwonych. Zgodnie ze swoim metabolizmem może być sklasyfikowana jako bilirubina sprzężona (skorelowana z bilirubiną bezpośrednią) i bilirubina niesprzężona (skorelowana z bilirubiną pośrednią).
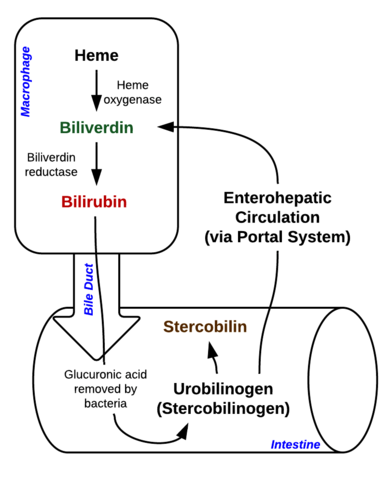
Obraz: „Breakdown of Heme in macrophages and intestine” autorstwa Johndheathcote. Licencja: CC BY-SA 3.0
Hyperbilirubinemię można zdefiniować jako podwyższone stężenie bilirubiny we krwi, które wyraża się w postaci żółtaczki (zwanej też icterus). Żółtaczka pojawia się, gdy żółte pigmenty bilirubiny odkładają się w skórze, twardówce, błonach śluzowych i innych tkankach, prowadząc do żółtawego przebarwienia.
Normalny zakres bilirubiny wynosi:
- Bilirubina całkowita w surowicy między 0,2 a 1,2 mg/dl.
- Bilirubina bezpośrednia w surowicy między 0.1 i 0,4 mg/dl.
Uważa się, że jest to hiperbilirubinemia, gdy:
- Bilirubina bezpośrednia w surowicy wynosi ≥ 1 mg/dl (jeżeli bilirubina całkowita wynosi < 5mg/dl).
- Bilirubina sprzężona w surowicy wynosi > 20% bilirubiny całkowitej (jeżeli bilirubina całkowita wynosi > 5mg/dl).
Etiologia pediatrycznej hiperbilirubinemii sprzężonej
U niektórych noworodków, zwłaszcza wcześniaków, niewielki wzrost bilirubiny może być uznany za normalny; jednak zawsze musi być badany, ponieważ wysoki poziom bilirubiny kosztem bilirubiny sprzężonej nigdy nie może być uznany za normalny.
Istnieje wiele chorób, które mogą powodować hiperbilirubinemię, ale można je ogólnie podzielić na dwie grupy: zaburzenia pozawątrobowe (przyczyny przedwątrobowe, takie jak hemoliza spowodowana wewnętrznymi wadami krwinek czerwonych lub przyczyny zewnętrzne prowadzące do pęknięcia krwinek czerwonych, lub przyczyny pozawątrobowe, w tym niedrożność dróg żółciowych) i zaburzenia wewnątrzwątrobowe (choroby wątroby).
Istnieje kilka stanów hiperbilirubinemii sprzężonej u dzieci :
- Infekcje: Wirusowe (WZW A-E, opryszczka, adenowirus, cytomegalowirus, HIV); bakteryjne (sepsa, zakażenia dróg moczowych), różyczka, gruźlica itp.
- Zaburzenia powodujące niedrożność dróg żółciowych, takie jak atrezja dróg żółciowych, perforacja dróg żółciowych, kamica żółciowa, itp
- Choroby układu wewnątrzwydzielniczego: niedoczynność tarczycy, dysplazja septooptyczna, itp.
- Genetyczne/metaboliczne: mukowiscydoza, zespół Alagille’a, alfa-1-antytrypsyna, itp.
- Zaburzenia magazynowania: Zespół Gauchera, choroby spichrzeniowe glikogenu, Nieman-Pick, itp.
- Ekspozycja na wiele leków, w tym ceftriakson, metotreksat, erytromycynę, tetracyklinę, itp.
Diagnostyka pediatrycznej hiperbilirubinemii sprzężonej
Historia
Aby postawić diagnozę hiperbilirubinemii u dzieci, bardzo ważne jest zebranie dobrego wywiadu klinicznego, w tym rodziców i bliskich krewnych, z uwzględnieniem patologicznych predyspozycji, ewentualnego pokrewieństwa między rodzicami, ewentualnych powikłań w czasie ciąży i porodu, ewentualnej ekspozycji na chorobę zakaźną przed lub po urodzeniu (m.in. zapalenie wątroby, malaria, cytomegalowirus, leptospiroza).
Obraz: „Noworodek poddawany fototerapii w leczeniu żółtaczki noworodkowej” autorstwa Marcina Garnka. Licencja: CC BY 3.0
Badanie
Badanie fizykalne jest bardzo ważne w diagnostyce hiperbilirubinemii. Głównym objawem jest żółtaczka lub żółtawe przebarwienie twardówek, błon śluzowych i skóry. Mocz dziecka staje się ciemny i, w zależności od przyczyny, kał może być biały.
Niezwykle ważne jest również wykonanie badania neurologicznego dziecka (odruchy i reakcja na bodźce zewnętrzne) w celu określenia schorzenia centralnego układu nerwowego (które jest znane jako kernicterus lub encefalopatia bilirubinowa). Wskazuje to na obecność ciężkiej hiperbilirubinemii.
Inne ważne parametry, które muszą być zbadane to parametry wzrostu, oznaki życiowe, szmery sercowe i oddechowe, badanie jamy brzusznej (w celu ustalenia nieprawidłowego wzrostu śledziony lub wątroby).
Badania laboratoryjne w pediatrycznej hiperbilirubinemii sprzężonej
Wymagane badania laboratoryjne to:
- Kompletna liczba krwinek, która jest niezbędna do badań przesiewowych w kierunku hemolizy.
- Poziomy aminotransferaz w surowicy są wykonywane w celu zbadania czynności wątroby. Obejmują one aminotransferazę asparaginianową i aminotransferazę alaninową.
- Serologiczne badania przesiewowe w kierunku wirusów.
- Fosfataza zasadowa, ponieważ jej wzrost może wskazywać na niedrożność dróg żółciowych.
- Transpeptydaza gamma-glutamylowa, której poziom może pomóc w odróżnieniu wątrobowego źródła podwyższonego ALP od innych przyczyn.
- Bilirubina frakcjonowana.
Innymi słowy, badania czynnościowe wątroby (LFT lub LF) są niezbędne do postawienia diagnozy i obejmują czas protrombinowy (PT/INR), czas częściowej tromboplastyny po aktywacji (aPTT), albuminy, bilirubinę (bezpośrednią i pośrednią) oraz transaminazy wątrobowe (AST i ALT). Biopsja i badania obrazowe są zwykle zarezerwowane dla przypadków z niejasnym rozpoznaniem lub wykluczają patologie obturacyjne.
Cholestatyczne przyczyny pediatrycznej sprzężonej hiperbilirubinemii
Atrezja dróg żółciowych
Atrezja dróg żółciowych jest kolejną bardzo częstą przyczyną sprzężonej hiperbilirubinemii. Pacjent może mieć całkowitą lub częściową niedrożność zewnątrzwątrobowego drzewa żółciowego w różnych miejscach, jednak najczęstszymi częściami dotkniętymi niedrożnością są przewód wątrobowy i przewód żółciowy wspólny. Częściowa niedrożność jest zwykle spowodowana zapaleniem włóknistym wzdłuż przewodu żółciowego.
To zapalenie włókniste jest spowodowane chorobą okołoporodową, zwykle infekcją wirusową, która wpływa na błonę śluzową przewodu żółciowego. Kiedy infekcja postępuje, układ immunologiczny odpowiada i dochodzi do pogrubienia nabłonka w błonie śluzowej, co prowadzi do niedrożności przewodu. W miarę upływu czasu w rozrastającej się błonie śluzowej dochodzi do stwardnienia i zwłóknienia, co powoduje całkowity zanik przewodu w pierwszym miesiącu życia i jego późniejsze powikłania.
W diagnostyce atrezji dróg żółciowych bardzo ważne jest również zebranie pełnego wywiadu klinicznego, przeprowadzenie dobrego badania fizykalnego i badań laboratoryjnych, jak wspomniano wcześniej; jednak w tym przypadku techniki obrazowe (takie jak ultrasonografia, badanie wątrobowo-żółciowe (HIDA) lub nawet cholangiogram śródoperacyjny) mogą być bardzo przydatne w rozpoznaniu choroby.
Żadne pierwotne leczenie medyczne nie jest skuteczne w korekcji atrezji dróg żółciowych, dlatego po potwierdzeniu rozpoznania konieczna jest interwencja chirurgiczna, a w wielu przypadkach sam zabieg może być diagnostyczny. Może być wykonany jako cholangiogram śródoperacyjny lub portoenterostomia Kasai. Przeszczepy wątroby są zarezerwowane dla ciężkich przypadków.
Torbiele żółciowe
Wreszcie, torbiele żółciowe, lepiej znane jako wrodzone poszerzenie dróg żółciowych, mogą również powodować sprzężoną hiperbilirubinemię. Najczęstszą wrodzoną nieprawidłowością torbieli żółciowych jest poszerzenie zewnątrzwątrobowych dróg żółciowych (1 na 100 000-150 000 noworodków). Objawy torbieli żółciowych mogą wystąpić w każdym wieku, jednak u niemowląt i dzieci charakteryzują się żółtaczką obturacyjną z bólami brzucha. Występują głównie u kobiet, a stosunek liczby kobiet do liczby mężczyzn wynosi około 3-4:1. Występują one częściej u niektórych ras azjatyckich.
Typy torbieli żółciowych: Początkowo opisano trzy typy torbieli żółciowych, ale Todani i współpracownicy dalej sklasyfikowali je w pięć typów, które są opisane poniżej:
- Typ I: Występuje poszerzenie przewodu żółciowego wspólnego. Może być ono torbielowate, ogniskowe lub stożkowate (obserwowane w 90-95% przypadków).
- Typ II: Widoczny jest tu uchyłek zewnątrzwątrobowej drogi żółciowej.
- Typ III: Charakteryzuje się występowaniem pęcherzyków żółciowych.
- Typ IV: Występuje w dwóch typach, Typ IV-A i Typ IV-B. Typ IV-A jest drugim najczęstszym typem i jest definiowany jako wewnątrzwątrobowe i zewnątrzwątrobowe poszerzenie dróg żółciowych. Typ IV-B obejmuje rzadką malformację licznych torbieli pozawątrobowych.
- Typ V: Ten typ jest również znany jako choroba Caroli, gdy jest związany z włóknieniem wątroby. Typ V obejmuje pojedyncze lub mnogie torbiele wewnątrzwątrobowe.
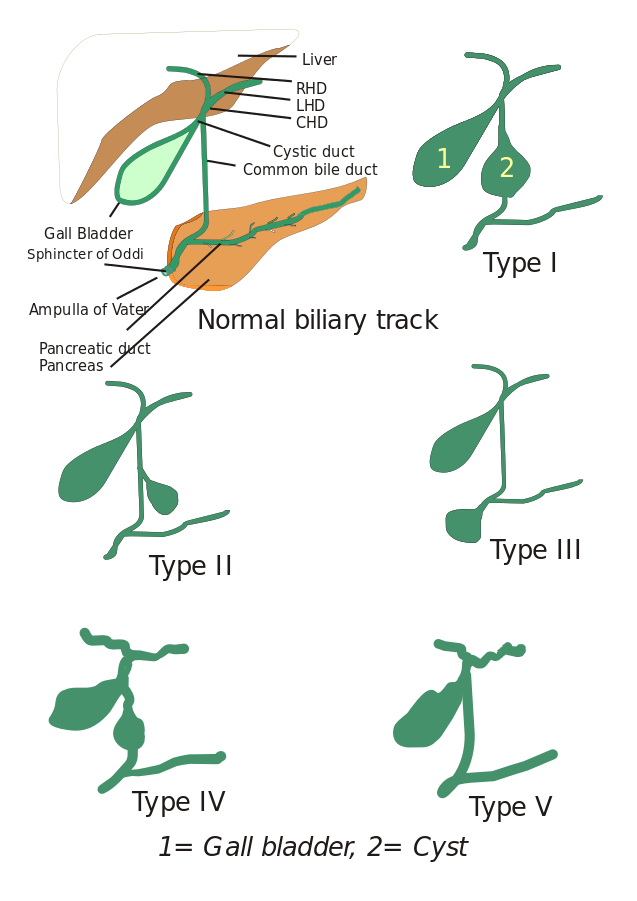
Obraz: „5 typów torbieli choledochalnych. Typ I: Rozszerzenie zewnątrzwątrobowego przewodu żółciowego; Typ II: Torbiel z przewodu żółciowego wspólnego (CBD); Typ III: Choledochocele lub rozszerzenie dystalnej części CBD, Typ IV: Rozszerzenie zarówno przewodu zewnątrzwątrobowego, jak i wewnątrzwątrobowego; Typ V: Choroba Caroli, Poszerzenie tylko przewodu wewnątrzwątrobowego. CHD: Wspólny przewód wątrobowy, LHD: Lewy przewód wątrobowy i RHD: Prawy przewód wątrobowy.” autorstwa I, Drriad. License: CC BY-SA 3.0
Powszechnie występuje u niemowląt w wieku od 1 do 3 miesięcy. Klinicznie objawia się gorączką, bólem w prawym górnym kwadrancie brzucha, acholicznymi lub bladymi stolcami i hepatomegalią, a obraz ten jest podobny do atrezji dróg żółciowych. W przypadku prenatalnego rozpoznania torbieli żółciowej, żółtaczka ujawnia się dopiero po 1-3 tygodniach od urodzenia. W przypadku późniejszego rozpoznania torbieli żółciowej, staje się ona klinicznie widoczna dopiero po 2 roku życia i może przebiegać z klasyczną triadą: ból brzucha, wyczuwalna masa brzuszna i żółtaczka, z których dwie występują u prawie 85% dzieci w momencie rozpoznania. Inne częste cechy to zapalenie dróg żółciowych, zapalenie trzustki i żółciowe zapalenie otrzewnej spowodowane pęknięciem torbieli.
Badania laboratoryjne, jak również badania obrazowe, są niezbędne do postawienia ostatecznej diagnozy. Badania laboratoryjne obejmują konwencjonalne testy czynności wątroby, morfologię krwi i profile krzepnięcia. Badania obrazowe obejmują między innymi ultrasonografię, tomografię komputerową, rezonans magnetyczny, cholangiopankreatografię rezonansu magnetycznego i endoskopową cholangiopankreatografię wsteczną.
Skanowanie ultrasonograficzne pomaga w rozpoznaniu torbieli żółciowych i ma bardzo wysoką swoistość 97% u dzieci, ale niską dokładność diagnostyczną w okresie przedporodowym; dlatego jest stosowane jako podstawowa metoda diagnostyki obrazowej w przypadku żółtaczki noworodków, która utrzymuje się dłużej niż 2 tygodnie po urodzeniu. Jest również przydatna w różnicowaniu między torbielami żółciowymi a atrezją dróg żółciowych.
Użycie tomografii komputerowej w diagnostyce torbieli żółciowych jest kontrowersyjne. Małe rozmiary torbieli lub choledochocele są przeoczone w tomografii komputerowej, ale są łatwo widoczne w cholangiopankreatografii rezonansu magnetycznego (MRCP). W okresie pooperacyjnym tomografia komputerowa może być lepsza niż MRCP w wykrywaniu położenia zespolenia żółciowo-jelitowego i w określaniu jego zwężenia.
Cholangiopankreatografia rezonansu magnetycznego (MRCP) jest lepsza w wykrywaniu i określaniu zmian chorobowych. Obecnie uważana jest za złoty standard w obrazowaniu torbieli żółciowych, jednak jej wadą jest brak możliwości dokładnego wykrycia anomalii w zespoleniu trzustkowo-żółciowym. Nie odgrywa to jednak istotnej roli w ustalaniu postępowania z chorym. Ponadto czułość MRCP jest większa u dorosłych niż u niemowląt.
Endoskopowa cholangiopankreatografia wsteczna (ECPW) jest najlepszym narzędziem do poznania anatomii dróg żółciowych i dlatego jest pomocna w diagnostyce torbieli żółciowych.
Leczenie obejmuje przede wszystkim interwencję chirurgiczną. W przeszłości w leczeniu chirurgicznym torbieli żółciowych stosowano procedurę drenażu wewnętrznego, znaną jako cysto-enterostomia (może to być albo cysto-duodenektomia, albo cysto-jejunostomia); jednak po operacji obserwowano różne powikłania, takie jak złośliwość w pozostałej torbieli, zapalenie trzustki i zapalenie dróg żółciowych; dlatego też z niej zrezygnowano.
Obecnie zalecanym postępowaniem chirurgicznym jest wycięcie torbieli, a następnie wykonanie hepaticojejunostomii Roux-en-Y lub choledochojejunostomii. Wycięcie torbieli zmniejsza częstość tworzenia się zwężeń w okresie pooperacyjnym. Sugerowane są również inne alternatywne rozwiązania, np. duodenotomia wątrobowa, aby w przypadku powikłań pooperacyjnych zespolenie było dostępne dla ECPW. Jednak stosowanie duodenotomii wątrobowej nie jest powszechnie akceptowane, ponieważ może ona powodować refluks żółciowy i zapalenie dróg żółciowych.
Po wycięciu torbieli konieczne jest sondowanie i obfite płukanie przewodów wewnątrzwątrobowych solą fizjologiczną w celu całkowitego usunięcia osadu i ewentualnych kamieni z przewodu. Ponadto, niedrożność może być obecna w proksymalnym układzie żółciowym, który można rozszerzyć. W związku z tym przed wycięciem torbieli obowiązkowe jest wykonanie cholangiografii śródoperacyjnej.
Defekty genetyczne dziecięcej hiperbilirubinemii sprzężonej
Istnieje wiele zespołów dziedzicznych związanych z hiperbilirubinemią i cholestazą wewnątrzwątrobową. Wiele z nich jest związanych z mutacjami genetycznymi, w tym genu SERPINAI (alfa 1-antytrypsyna), JAG1 (powodującego zespół Alagille’a), ATP8B1 (znanego również jako FIC1), ABCB11 (pompa eksportu soli żółciowych), MDR3 (ABCB4) i MRP2 (powodującego zespół Dubina-Johnsona).
1) Zespół Alagille’a
Zespół Alagille’a jest dziedziczonym autosomalnie dominująco zaburzeniem genetycznym, które może dotyczyć wielu części ciała. Mutacja występuje w krótkim ramieniu chromosomu 20 (20p12). Jedna na każde 20 lub 30 mutacji pojawia się de novo. Jednym z najbardziej dotkniętych narządów w zespole Alagille’a jest wątroba i drogi żółciowe. Te wady rozwojowe dróg żółciowych powodują gromadzenie się żółci w wątrobie, czyli cholestazę, która powoduje bliznowacenie wątroby, zmieniając w ten sposób jej funkcjonowanie.
Objawy wynikające z uszkodzenia wątroby w zespole Alagille’a mogą obejmować żółtaczkę, swędzenie skóry i ksantomy. Jednak uszkodzeniu ulega również serce, a u pacjenta może wystąpić zwężenie tętnicy płucnej. W zwężeniu tętnicy płucnej dochodzi do upośledzenia przepływu krwi z serca do płuc. Mogą również współistnieć wrodzone choroby serca, takie jak ubytki w przegrodzie międzykomorowej, tetralogia Fallota itp. Mózg, rdzeń kręgowy, nerki i naczynia krwionośne mogą być również dotknięte.
Zespół Alagille’a ma charakterystyczne rysy twarzy. Pacjent ma zazwyczaj trójkątną twarz, szerokie i wydatne czoło, szeroki mostek nosowy, głęboko osadzone oczy oraz mały, spiczasty podbródek. Rozpoznanie jest trudne i wymaga co najmniej 3 typowych cech fizycznych, stwierdzenia niedrożności dróg żółciowych (cholestazy) oraz wykonania biopsji wątroby.
Leczenie zależy od ciężkości choroby w zespole Alagille’a. W przypadku łagodnej choroby podaje się kwas ursodeoksycholowy w celu ułatwienia przepływu żółci oraz leki przeciwhistaminowe, takie jak difenhydramina, w celu opanowania świądu. W ciężkich przypadkach może być konieczny przeszczep wątroby. Stwierdzono, że pomocna jest suplementacja witamin, zwłaszcza witamin A, D, E i K. Korzystna jest również suplementacja cynku. Pacjenci ci nie są w stanie wchłonąć tych witamin i dlatego suplementacja pomaga w ułatwieniu optymalnego rozwoju pacjenta.
2) Zespół Dubina-Johnsona
Zespół ten charakteryzuje się izolowaną sprzężoną przewlekłą hiperbilirubinemią, bez dowodów hemolizy, spowodowaną defektem eliminacji sprzężonej bilirubiny w żółci (cholestaza wewnątrzwątrobowa). Jest ona spowodowana mutacją w genie MRP2. Jest to łagodny stan, który nie wymaga specyficznego leczenia.
Zespół Rotora
Jest to rzadkie zaburzenie charakteryzujące się przewlekłą sprzężoną i niesprzężoną hiperbilirubinemią bez hemolizy. Dziedziczenie genetyczne nadal nie jest znane. Występuje z powodu defektu magazynowania bilirubiny sprzężonej w wątrobie, która przecieka do osocza powodując hiperbilirubinemię.
Ucz się do szkoły medycznej i rad nadzorczych z Lecturio.
- USMLE Step 1
- USMLE Step 2
- COMLEX Level 1
- COMLEX Level 2
- ENARM
- NEET