
Imagine: „Bebeluș în secția de terapie intensivă neonatală.” de Jacoplane (ei bine, părinții lui, oricum) – Operă proprie. Licență: CC BY-SA 3.0
- Introducere
- Etiologia hiperbilirubinemiei conjugate pediatrice
- Diagnosticul hiperbilirubinemiei conjugate pediatrice
- Istoric
- Examinare
- Testele de laborator ale hiperbilirubinemiei conjugate pediatrice
- Cauze colestatice ale hiperbilitubinemiei conjugate pediatrice
- Atrezie biliară
- Cisturile coledocale
- Defecte genetice ale hiperbilirubinemiei conjugate pediatrice
- 1) Sindromul Alagille
- 2) Sindromul Dubin-Johnson
- Sindromul Rotor
Introducere
Bilirubina este un pigment (tetrapirrol) care se formează în calea catabolică normală din gruparea heme. Este, așadar, un produs rezidual al distrugerii globulelor roșii mature. În funcție de metabolismul său, poate fi clasificată în bilirubină conjugată (corelată cu bilirubina directă) și bilirubină neconjugată (corelată cu bilirubina indirectă).
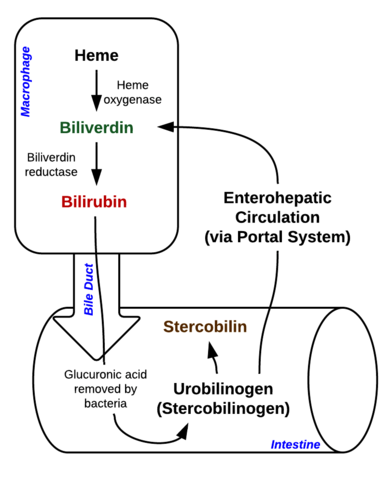
Imagine: „Descompunerea hemului în macrofage și intestin” de Johndheathcote. Licență: CC BY-SA 3.0
Hiperbilirubinemia poate fi definită ca o concentrație crescută de bilirubină în sânge, care se exprimă sub formă de icter (numit și icter). Icterul apare atunci când pigmenții galbeni ai bilirubinei se depun în piele, scleră, mucoase și alte țesuturi, ducând la o decolorare gălbuie.
Intervalul normal al bilirubinei este:
- Bilirubina totală serică între 0,2 și 1,2 mg/dL.
- Bilirubina directă serică între 0.1 și 0,4 mg/dL.
Se consideră hiperbilirubinemie când:
- Bilirubina directă serică este ≥ 1 mg/dL (dacă bilirubina totală este < 5mg/dL).
- Bilirubina conjugată serică este > 20% din bilirubina totală (dacă bilirubina totală este > 5mg/dL).
Etiologia hiperbilirubinemiei conjugate pediatrice
La unii nou-născuți, în special la prematuri, o ușoară creștere a bilirubinei poate fi considerată normală; cu toate acestea, ea trebuie întotdeauna studiată, deoarece un nivel ridicat de bilirubină în detrimentul bilirubinei conjugate nu poate fi niciodată considerat normal.
Există multe boli care pot cauza hiperbilirubinemie, dar acestea pot fi clasificate în linii mari în două grupe: afecțiuni extrahepatice (cauze prehepatice, cum ar fi hemoliza datorată unor defecte intrinseci ale globulelor roșii sau cauze extrinseci care duc la ruperea globulelor roșii, sau cauze post-hepatice, inclusiv obstrucția pasajului biliar) și afecțiuni intrahepatice (boli hepatice).
Există mai multe afecțiuni ale hiperbilirubinemiei conjugate la copii :
- Infecții: Virale (hepatita A-E, herpes, adenovirus, citomegalovirus, HIV); bacteriene (sepsis, infecții urinare), rubeolă, tuberculoză etc.
- Dezordini care cauzează obstrucția căii biliare, cum ar fi atrezia biliară, perforarea căii biliare, colelitiaza, etc
- Boli ale sistemului endocrin: hipotiroidism, displazia septoptică, etc.
- Genetice/metabolice: fibroza chistică, sindromul Alagille, alfa-1-antitripsina, etc.
- Dezordini de stocare: Sindromul Gaucher, boli de stocare a glicogenului, Nieman-Pick, etc.
- Expunere la multe medicamente, inclusiv ceftriaxonă, metotrexat, eritromicină, tetraciclină, etc.
Diagnosticul hiperbilirubinemiei conjugate pediatrice
Istoric
Pentru a pune diagnosticul de hiperbilirubinemie la copii, este foarte important să se facă o bună anamneză clinică, incluzând antecedentele patologice ale părinților și rudelor apropiate, posibila existență a consangvinității între părinți, eventualele complicații în timpul sarcinii și al nașterii, posibila expunere la o boală infecțioasă înainte sau după naștere (cum ar fi hepatita, malaria, citomegalovirusul și leptospiroza), printre altele.
Imagine: „Bebeluș nou-născut supus fototerapiei pentru tratarea icterului neonatal” de Martin Pot. Licență: CC BY 3.0
Examinare
Examinarea fizică este foarte importantă în diagnosticul hiperbilirubinemiei. Semnul principal este icterul sau colorația gălbuie a sclierelor, a mucoaselor și a pielii. Urina copilului devine închisă la culoare și, în funcție de cauză, materiile fecale pot fi albe.
De asemenea, este extrem de important să se efectueze un examen neurologic al copilului (reflexele și răspunsul la stimuli externi) pentru a determina afecțiunea sistemului nervos central (care este cunoscută sub numele de kernicterus sau encefalopatie bilirubinică). Aceasta indică prezența unei hiperbilirubinemii severe.
Alți parametri importanți care trebuie examinați sunt parametrii de creștere, semnele vitale, suflul cardiac și respirator, examenul abdominal (pentru a determina creșterea anormală a splinei sau a ficatului).
Testele de laborator ale hiperbilirubinemiei conjugate pediatrice
Testele de laborator necesare sunt:
- Contul complet al celulelor sanguine, care este necesar pentru depistarea hemolizei.
- Nivelurile aminotransferazelor serice se fac pentru a studia funcția ficatului. Acestea includ aspartat aminotransferaza și alanin aminotransferaza.
- Serologic pentru depistarea virusurilor.
- Fosfataza alcalină deoarece o creștere a acesteia poate indica obstrucția căilor biliare.
- Gamma-glutamil transpeptidaza ale cărei niveluri pot ajuta la diferențierea unei surse hepatice a ALP crescute de alte cauze.
- Bilirubina fracționată.
În alți termeni, testele funcției hepatice (LFT sau LF) sunt necesare pentru diagnostic și includ timpul de protrombină (PT/INR), timpul de tromboplastină parțială activată (aPTT), albumina, bilirubina (directă și indirectă) și transaminazele hepatice (AST și ALT). Biopsia și studiile imagistice sunt de obicei rezervate acelor cazuri cu un diagnostic neclar sau se discută patologii obstructive.
Cauze colestatice ale hiperbilitubinemiei conjugate pediatrice
Atrezie biliară
Atrezia biliară este o altă cauză foarte frecventă de hiperbilirubinemie conjugată. Pacientul poate avea o obstrucție completă sau parțială a arborelui biliar extrahepatic în diferite puncte; cu toate acestea, cele mai frecvente părți afectate sunt canalul hepatic și canalul biliar comun. Obstrucția parțială este de obicei cauzată de o fibroinflamare de-a lungul canalului biliar.
Această fibroinflamare se datorează unei boli perinatale, de obicei o infecție virală care afectează mucoasa canalului biliar. Când infecția progresează, sistemul imunologic răspunde și se produce o îngroșare epitelială a mucoasei care duce la obstrucția ductului. Pe măsură ce timpul avansează, mucoasa proliferantă dezvoltă scleroză și fibroză, provocând atrezia totală a canalului în prima lună de viață și complicațiile ulterioare.
În diagnosticul de atrezie biliară, este, de asemenea, foarte important să se facă o anamneză clinică completă, să se efectueze un bun examen fizic și teste de laborator, așa cum s-a menționat anterior; totuși, în acest caz, tehnicile imagistice (cum ar fi ecografia, scanarea hepatobiliară (HIDA) sau chiar o colangiogramă intraoperatorie) pot fi foarte utile în diagnosticul bolii.
Niciun tratament medical primar nu este eficient în corectarea atreziei biliare; prin urmare, odată ce diagnosticul este confirmat, este necesară intervenția chirurgicală și, în multe cazuri, intervenția chirurgicală în sine poate fi diagnostică. Aceasta se poate face sub forma unei colangiograme intraoperatorii sau a unei portoenterostomii Kasai. Transplantul de ficat este rezervat pentru cazurile severe.
Cisturile coledocale
În cele din urmă, chisturile coledocale, mai bine cunoscute sub denumirea de dilatare congenitală a căilor biliare, pot provoca, de asemenea, hiperbilirubinemie conjugată. Cea mai frecventă anomalie congenitală a chisturilor coledociene este dilatarea canalului biliar extrahepatic (1 la fiecare 100.000-150.000 de nou-născuți). Simptomele chisturilor coledocale se pot prezenta la orice vârstă; cu toate acestea, ele sunt caracteristice cu icter obstructiv împreună cu dureri abdominale la sugari și copii. Au o predilecție feminină, cu un raport femeie/bărbat de aproximativ 3-4:1. Sunt mai frecvente la anumite rase asiatice.
Tipuri de chisturi coledociene: Inițial, au fost descrise trei tipuri de chisturi coledociene, dar Todani și asociații le-au clasificat ulterior în cinci tipuri care sunt descrise mai jos:
- Tipul I: Există o dilatare a canalului biliar comun. Aceasta poate fi chistică, focală sau fusiformă (observată în 90-95% din cazuri).
- Tipul II: Aici se observă un diverticul al canalului biliar extrahepatic.
- Tipul III: Caracterizat de coledoccele.
- Tipul IV: Este de două tipuri, tipul IV-A și tipul IV-B. Tipul IV-A este al doilea cel mai frecvent tip și este definit ca o dilatare atât intrahepatică cât și extrahepatică a arborelui biliar. Tipul IV-B implică malformația rară a chisturilor multiple extrahepatice.
- Tipul V: Acest tip este cunoscut și sub numele de boala Caroli atunci când este asociat cu fibroza hepatică. Tipul V include chisturi intrahepatice unice sau multiple.
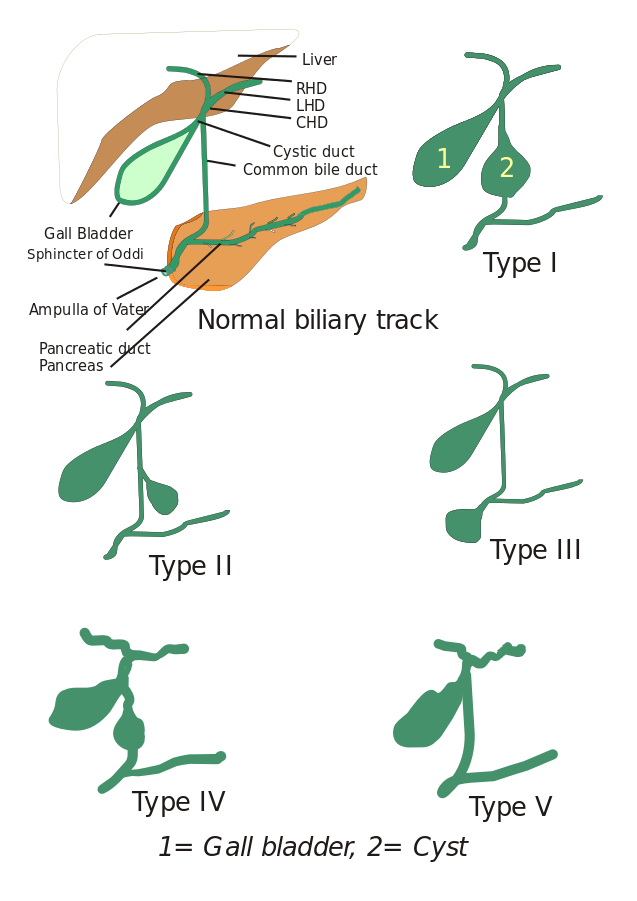
Imagine: „5 tipuri de chisturi coledocale. Tipul I: Dilatarea canalului biliar extrahepatic; Tipul II: Chist din canalul biliar comun (CBD); Tipul III: Coledococele sau dilatarea părții distale a CBD, tipul IV: Dilatarea atât a canalului extrahepatic cât și a celui intrahepatic; Tipul V: Boala Caroli, dilatarea doar a canalului intrahepatic. CHD: Ductul hepatic comun, LHD: Ductul hepatic stâng și RHD: Ductul hepatic drept.” De I, Drriad. Licență: CC BY-SA 3.0
Se întâlnește frecvent la sugari cu vârsta cuprinsă între 1 și 3 luni. Din punct de vedere clinic, se prezintă cu febră, durere în cadranul superior drept al abdomenului, scaune acholice sau palide și hepatomegalie, iar acest tablou este similar cu atrezia biliară. În cazul diagnosticului prenatal al chistului coledoc, icterul nu este evident decât la 1 până la 3 săptămâni de la naștere. În cazul în care chistul coledoc se prezintă mai târziu, acesta nu devine clinic evident decât după vârsta de 2 ani și se poate prezenta cu o triadă clasică de durere abdominală, masă abdominală palpabilă și icter; dintre care două sunt întâlnite la aproape 85% dintre copii în momentul prezentării. Alte caracteristici comune sunt colangita, pancreatita și peritonita biliară datorată rupturii chistului.
Investigațiile de laborator, precum și imagistica, sunt necesare pentru un diagnostic definitiv. Investigațiile de laborator includ testele convenționale ale funcțiilor hepatice, hemograma și profilele de coagulare. Studiile imagistice includ ecografia, tomografia computerizată, imagistica prin rezonanță magnetică, colangiopancreatografia prin rezonanță magnetică și colangiopancreatografia endoscopică retrogradă, printre altele.
Ecografia ajută la diagnosticarea chisturilor coledocale și are o specificitate foarte mare, de 97% la copii, dar o acuratețe diagnostică scăzută în perioada antenatală; astfel, este utilizată ca modalitate imagistică de diagnostic primar pentru icterul neonatal care persistă mai mult de 2 săptămâni după naștere. De asemenea, este utilă în diferențierea dintre chisturile coledocale și atrezia biliară.
Utilizarea tomografiei computerizate pentru diagnosticarea chisturilor coledocale este controversată. Chisturile de dimensiuni mici sau coledococele sunt ratate la tomografia computerizată, dar sunt ușor de observat la colangiopancreatografia prin rezonanță magnetică (MRCP). Tomografia computerizată poate fi mai bună decât MRCP în perioada postoperatorie pentru a detecta localizarea anastomozei biliaro-enterice și pentru a defini orice stenoză a acesteia.
Colangiopancreatografia prin rezonanță magnetică (MRCP) este superioară la detectarea și definirea leziunilor. În prezent, este considerată standardul de aur în imagistica chisturilor coledocale; cu toate acestea, are dezavantajul de a nu putea detecta cu precizie orice unificare pancreaticobiliară anormală. Cu toate acestea, acest lucru nu joacă un rol major în determinarea managementului pacientului. În plus, sensibilitatea MRCP este mai mare la adulți decât la sugari.
Colangiopancreatografia endoscopică retrogradă (ERCP) este cel mai bun instrument pentru cunoașterea anatomiei biliare și este astfel utilă în diagnosticarea chisturilor coledociene.
Tratamentul include în primul rând intervenția chirurgicală. O procedură de drenaj intern cunoscută sub numele de chist-enterostomie (care ar putea fi fie chist-duodenectomie, fie chist-jejunostomie) a fost efectuată în trecut pentru tratamentul chirurgical al chisturilor coledociene; cu toate acestea, au fost observate diverse complicații postoperatorii, cum ar fi malignitate în chistul rămas, pancreatită și colangită; astfel, acestea au fost abandonate.
În prezent, procedura chirurgicală recomandată este excizia chistului și apoi hepatico-jejunostomia Roux-en-Y sau coledocojejunostomia. Datorită exciziei chisturilor, există o reducere a incidenței formării de stricturi postoperator. Au fost sugerate și alte alternative, de exemplu duodenotomia hepatică, astfel încât anastomoza să fie accesibilă la ERCP în caz de complicații postoperatorii. Cu toate acestea, utilizarea duodenotomiei hepatice nu a fost acceptată pe scară largă, deoarece poate provoca reflux biliar și colangită.
Post-excizia chistului, este necesară sondarea și spălarea abundentă a canalelor intrahepatice cu soluție salină pentru a elimina complet nămolul și eventualele pietre din sistemul ductal. În plus, obstrucția poate fi prezentă în sistemul biliar proximal care poate fi dilatat. În consecință, înainte de excizia chistului, este obligatorie o colangiografie intraoperatorie.
Defecte genetice ale hiperbilirubinemiei conjugate pediatrice
Există o mulțime de sindroame ereditare legate de hiperbilirubinemie și colestază intrahepatică. Multe dintre ele sunt legate de mutații genetice, inclusiv gena SERPINAI (alfa 1- antitripsină), JAG1 (care cauzează sindromul Alagille), ATP8B1 (cunoscută și sub numele de FIC1), ABCB11 (pompa de export a sărurilor biliare ), MDR3 (ABCB4) și MRP2 (care cauzează sindromul Dubin-Johnson).
1) Sindromul Alagille
Sindromul Alagille este o tulburare genetică autozomal dominantă care poate afecta multe părți ale corpului. Mutația apare în brațul scurt al cromozomului 20 (20p12). Una din 20 sau 30 de mutații apare de novo. Unul dintre cele mai afectate organe în sindromul Alagille este ficatul și canalele biliare. Aceste malformații ale canalelor biliare cauzează acumularea de bilă în ficat, adică colestaza, care provoacă cicatrizarea ficatului, alterând astfel funcționarea acestuia.
Semnele și simptomele care decurg din afectarea ficatului în sindromul Alagille pot include icter, mâncărimi ale pielii și xantome. Cu toate acestea, inima este, de asemenea, afectată și pacientul poate avea stenoză pulmonară. În stenoza pulmonară, există o afectare a fluxului sanguin dinspre inimă spre plămâni. Pot exista, de asemenea, boli cardiace congenitale coexistente, cum ar fi defectele septale ventriculare, tetralogia lui Fallot etc. Creierul, măduva spinării, rinichii și vasele de sânge pot fi, de asemenea, afectate.
Sindromul Alagille are trăsături faciale caracteristice. Pacientul are, de obicei, o față triunghiulară, o frunte largă și proeminentă, o punte nazală largă, ochi adânciți și o bărbie mică și ascuțită. Diagnosticul este dificil și necesită cel puțin 3 dintre trăsăturile fizice tipice, evidențierea obstrucției canalelor biliare (colestază) și o biopsie hepatică.
Tratamentul este dependent de severitatea bolii în sindromul Alagille. În cazul bolii ușoare, se administrează acid ursodeoxicolic pentru a facilita fluxul biliar și se administrează antihistaminice precum difenhidramina pentru a controla pruritul. Pentru cazurile severe, poate fi necesar un transplant de ficat. Suplimentarea cu vitamine s-a dovedit a fi utilă, în special cu vitaminele A, D, E și K. Suplimentarea cu zinc este, de asemenea, benefică. Acești pacienți au o incapacitate de a absorbi aceste vitamine și astfel, suplimentarea ajută la facilitarea creșterii optime a pacientului.
2) Sindromul Dubin-Johnson
Acest sindrom se caracterizează prin hiperbilirubinemie cronică conjugată izolată, fără dovezi de hemoliză, datorită unui defect în eliminarea bilirubinei conjugate în bilă (colestază intrahepatică). Este cauzată de o mutație în gena MRP2. Este o afecțiune benignă care nu necesită tratament specific.
Sindromul Rotor
Este o afecțiune rară caracterizată prin hiperbilirubinemie conjugată și neconjugată cronică fără hemoliză. Moștenirea genetică nu este încă cunoscută. Apare din cauza unui defect de stocare a bilirubinei conjugate în ficat, care se scurge în plasmă provocând hiperbilirubinemie.
Studiați pentru școala medicală și comisii cu Lecturio.
- USMLE Step 1
- USMLE Step 2
- COMLEX Level 1
- COMLEX Level 2
- ENARMARE
- NEET
.