
Image : « Bébé dans l’unité de soins intensifs néonatals » par Jacoplane (enfin, ses parents en tout cas) – Travail propre. Licence : CC BY-SA 3.0
- Introduction
- Etiologie de l’hyperbilirubinémie conjuguée pédiatrique
- Diagnostic de l’hyperbilirubinémie conjuguée pédiatrique
- Histoire
- Examen
- Les examens de laboratoire de l’hyperbilirubinémie conjuguée pédiatrique
- Causes cholestatiques de l’hyperbilirubinémie conjuguée pédiatrique
- Atrésie biliaire
- Kystes cholédociens
- Défauts génétiques de l’hyperbilirubinémie conjuguée pédiatrique
- 1) Syndrome d’Alagille
- 2) Syndrome de Dubin-Johnson
- Syndrome de Rotor
Introduction
La bilirubine est un pigment (tétrapyrrole) qui se forme dans la voie normale du catabolisme à partir du groupe hème. Elle est donc un déchet de la destruction des globules rouges matures. Selon son métabolisme, elle peut être classée en bilirubine conjuguée (corrélée à la bilirubine directe) et non conjuguée (corrélée à la bilirubine indirecte).
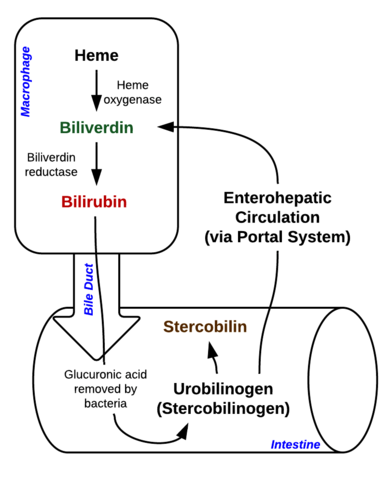
Image : « Décomposition de l’hème dans les macrophages et l’intestin » par Johndheathcote. Licence : CC BY-SA 3.0
L’hyperbilirubinémie peut être définie comme une concentration élevée de bilirubine dans le sang, qui s’exprime sous la forme d’un ictère (également appelé icterus). L’ictère apparaît lorsque les pigments jaunes de la bilirubine se déposent dans la peau, la sclérotique, les muqueuses et d’autres tissus, entraînant une coloration jaunâtre.
La plage normale de la bilirubine est :
- Bilirubine totale sérique entre 0,2 et 1,2 mg/dL.
- Bilirubine directe sérique entre 0.1 et 0,4 mg/dL.
On parle d’hyperbilirubinémie lorsque :
- La bilirubine directe sérique est ≥ 1 mg/dL (si la bilirubine totale est < 5mg/dL).
- La bilirubine conjuguée sérique est > 20% de la bilirubine totale (si la bilirubine totale est > 5mg/dL).
Etiologie de l’hyperbilirubinémie conjuguée pédiatrique
Chez certains nouveau-nés, notamment les prématurés, une légère augmentation de la bilirubine peut être considérée comme normale ; cependant, elle doit toujours être étudiée car un taux élevé de bilirubine aux dépens de la bilirubine conjuguée ne peut jamais être considéré comme normal.
Il existe de nombreuses maladies susceptibles de provoquer une hyperbilirubinémie, mais elles peuvent être globalement classées en deux groupes : les troubles extrahépatiques (causes préhépatiques comme l’hémolyse due à des défauts intrinsèques des globules rouges ou à des causes extrinsèques entraînant la rupture des globules rouges, ou causes post-hépatiques, notamment l’obstruction du passage biliaire) et les troubles intrahépatiques (maladies du foie).
Il existe plusieurs conditions d’hyperbilirubinémie conjuguée chez l’enfant :
- Infections : Virales (hépatites A-E, herpès, adénovirus, cytomégalovirus, VIH) ; bactériennes (septicémie, infections urinaires), rubéole, tuberculose, etc.
- Maladies entraînant une obstruction du canal biliaire telles que l’atrésie biliaire, la perforation du canal biliaire, la cholélithiase, etc
- Maladies du système endocrinien : hypothyroïdie, dysplasie septooptique, etc.
- Génétique/métabolique : mucoviscidose, syndrome d’Alagille, alpha-1-antitrypsine, etc.
- Troubles de stockage : Syndrome de Gaucher, maladies de stockage du glycogène, Nieman-Pick, etc.
- Exposition à de nombreux médicaments dont la ceftriaxone, le méthotrexate, l’érythromycine, la tétracycline, etc.
Diagnostic de l’hyperbilirubinémie conjuguée pédiatrique
Histoire
Pour poser un diagnostic d’hyperbilirubinémie chez l’enfant, il est très important de réaliser une bonne histoire clinique, incluant les antécédents pathologiques des parents et des proches, l’existence éventuelle d’une consanguinité entre les parents, d’éventuelles complications pendant la grossesse et l’accouchement, une éventuelle exposition à une maladie infectieuse avant ou après la naissance (comme l’hépatite, le paludisme, le cytomégalovirus et la leptospirose) entre autres.
Image : « Nouveau-né subissant une photothérapie pour traiter la jaunisse néonatale » par Martin Pot. Licence : CC BY 3.0
Examen
L’examen physique est très important dans le diagnostic de l’hyperbilirubinémie. Le principal signe est la jaunisse ou la coloration jaunâtre des sclérotiques, des muqueuses et de la peau. L’urine du bébé devient foncée et, selon la cause, les selles peuvent être blanches.
Il est également extrêmement important d’effectuer un examen neurologique du bébé (réflexes et réponse aux stimuli externes) afin de déterminer l’affection du système nerveux central (que l’on appelle ictère nucléaire ou encéphalopathie bilirubinique). Cela indique la présence d’une hyperbilirubinémie sévère.
Les autres paramètres importants qui doivent être examinés sont les paramètres de croissance, les signes vitaux, les souffles cardiaques et respiratoires, l’examen abdominal (afin de déterminer la croissance anormale de la rate ou du foie).
Les examens de laboratoire de l’hyperbilirubinémie conjuguée pédiatrique
Les examens de laboratoire nécessaires sont :
- La numération globulaire complète qui est nécessaire pour le dépistage de l’hémolyse.
- Les taux d’aminotransférases sériques sont effectués pour étudier la fonction du foie. Elles comprennent l’aspartate aminotransférase et l’alanine aminotransférase .
- Dépistage sérologique des virus.
- Phosphatase alcaline car son élévation peut indiquer une obstruction des voies biliaires.
- Gamma-glutamyl transpeptidase dont le taux peut aider à différencier une source hépatique de l’ALP élevée d’autres causes.
- Birubine fractionnée.
En d’autres termes, les tests de la fonction hépatique (LFT ou LF) sont nécessaires au diagnostic et ils comprennent le temps de prothrombine (PT/INR), le temps de thromboplastine partielle activée (aPTT), l’albumine, la bilirubine (directe et indirecte) et les transaminases hépatiques (AST et ALT). La biopsie et les études d’imagerie sont généralement réservées aux cas dont le diagnostic n’est pas clair ou écartent les pathologies obstructives.
Causes cholestatiques de l’hyperbilirubinémie conjuguée pédiatrique
Atrésie biliaire
L’atrésie biliaire est une autre cause très fréquente d’hyperbilirubinémie conjuguée. Le patient peut présenter une obstruction complète ou partielle de l’arbre biliaire extra-hépatique en différents points ; cependant, les parties les plus fréquemment touchées sont le canal hépatique et le canal biliaire commun. L’obstruction partielle est généralement causée par une fibro-inflammation le long du canal biliaire.
Cette fibro-inflammation est due à une maladie périnatale, généralement une infection virale qui affecte la muqueuse du canal biliaire. Lorsque l’infection progresse, le système immunologique réagit et il se produit un épaississement épithélial de la muqueuse conduisant à l’obstruction du canal. Avec le temps, la muqueuse en prolifération développe une sclérose et une fibrose, provoquant une atrésie totale du canal au cours du premier mois de vie et ses complications ultérieures.
Dans le diagnostic de l’atrésie des voies biliaires, il est également très important de prendre une histoire clinique complète, de réaliser un bon examen physique et des tests de laboratoire comme mentionné précédemment ; cependant, dans ce cas, les techniques d’imagerie (comme l’échographie, la scintigraphie hépatobiliaire (HIDA) ou même un cholangiogramme peropératoire) peuvent être très utiles dans le diagnostic de la maladie.
Aucun traitement médical primaire n’est efficace pour corriger l’atrésie des voies biliaires ; par conséquent, une fois le diagnostic confirmé, une intervention chirurgicale est nécessaire et, dans de nombreux cas, la chirurgie elle-même peut être diagnostique. Elle peut prendre la forme d’une cholangiographie peropératoire ou d’une portoentérostomie de Kasai. Les transplantations hépatiques sont réservées aux cas graves.
Kystes cholédociens
Enfin, les kystes cholédociens, plus connus sous le nom de dilatation congénitale des voies biliaires, peuvent également provoquer une hyperbilirubinémie conjuguée. L’anomalie congénitale la plus courante des kystes cholédociens est l’élargissement du canal biliaire extrahépatique (1 cas sur 100 000 à 150 000 nouveau-nés). Les symptômes des kystes cholédociens peuvent se manifester à tout âge ; toutefois, ils se caractérisent par un ictère obstructif accompagné de douleurs abdominales chez les nourrissons et les enfants. Ils ont une prédilection pour les femmes, avec un rapport femmes/hommes d’environ 3-4:1. Ils sont plus fréquents dans certaines races asiatiques.
Types de kystes cholédociens : Initialement, trois types de kystes cholédociens ont été décrits mais Todani et associés les ont ensuite classés en cinq types qui sont décrits ci-dessous :
- Type I : Il y a une dilatation du canal cholédoque. Elle peut être kystique, focale ou fusiforme (observée dans 90 à 95 % des cas).
- Type II : On observe ici un diverticule du canal biliaire extrahépatique.
- Type III : Caractérisé par des cholédocèles.
- Types IV : Il est de deux types, le type IV-A et le type IV-B. Le type IV-A est le deuxième type le plus fréquent et se définit par une dilatation à la fois intrahépatique et extrahépatique de l’arbre biliaire. Le type IV-B implique la malformation rare de multiples kystes extrahépatiques.
- Type V : Ce type est également connu sous le nom de maladie de Caroli lorsqu’il est associé à une fibrose hépatique. Le type V comprend des kystes intrahépatiques uniques ou multiples.
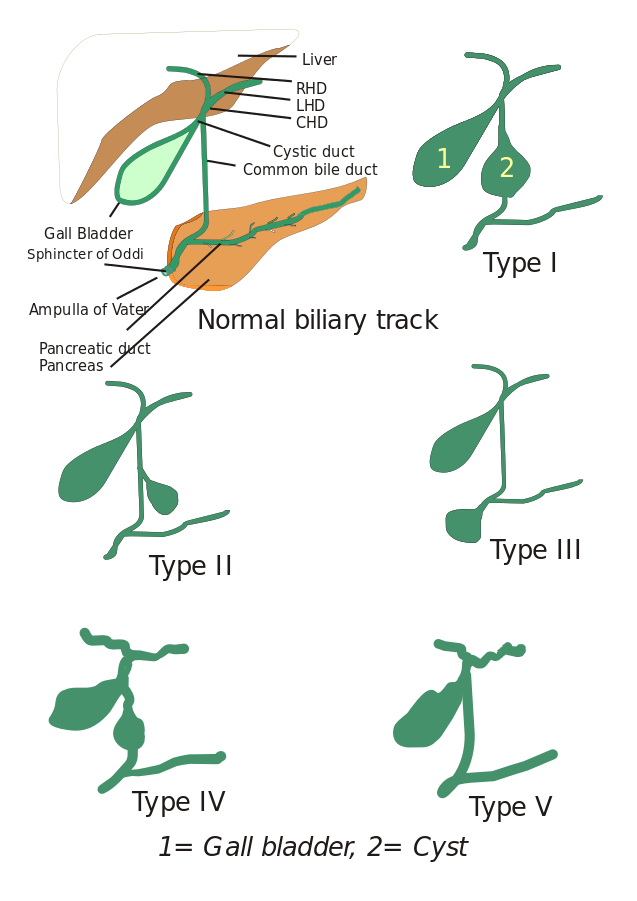
Image : « 5 types de kystes cholédociens. Type I : Dilatation du canal biliaire extrahépatique ; Type II : Kyste du canal biliaire commun (CBD) ; Type III : Cholédocèle ou dilatation de la partie distale du CBD, type IV : Dilatation du canal extrahépatique et intrahépatique ; Type V : Maladie de Caroli, Dilatation du canal intrahépatique uniquement. CHD : Conduit hépatique commun, LHD : Conduit hépatique gauche et RHD : Conduit hépatique droit. » Par moi, Drriad. Licence : CC BY-SA 3.0
Il est fréquemment observé chez les nourrissons âgés de 1 à 3 mois. Cliniquement, ils présentent de la fièvre, une douleur dans le quadrant supérieur droit de l’abdomen, des selles acholiques ou pâles et une hépatomégalie et ce tableau est similaire à celui de l’atrésie des voies biliaires. Dans le cas d’un diagnostic prénatal de kyste cholédoque, la jaunisse n’est apparente que 1 à 3 semaines après la naissance. Lorsque le kyste cholédoque se présente plus tard, il ne devient cliniquement évident qu’après l’âge de 2 ans et peut se présenter sous la forme d’une triade classique de douleurs abdominales, d’une masse abdominale palpable et d’un ictère, dont deux sont retrouvés chez près de 85 % des enfants au moment de la présentation. D’autres caractéristiques communes sont la cholangite, la pancréatite et la péritonite biliaire due à la rupture du kyste.
Les examens de laboratoire, ainsi que l’imagerie, sont nécessaires pour un diagnostic définitif. Les examens de laboratoire comprennent les tests conventionnels des fonctions hépatiques, la NFS et les profils de coagulation. Les études d’imagerie comprennent l’échographie, la tomodensitométrie, l’imagerie par résonance magnétique, la cholangiopancréatographie par résonance magnétique et la cholangiopancréatographie rétrograde endoscopique, entre autres.
L’échographie aide au diagnostic des kystes cholédociens et elle a une spécificité très élevée de 97% chez les enfants, mais une faible précision diagnostique en période anténatale ; elle est donc utilisée comme modalité d’imagerie diagnostique primaire pour l’ictère néonatal qui persiste plus de 2 semaines après la naissance. Elle est également utile pour différencier les kystes cholédociens de l’atrésie biliaire.
L’utilisation de la tomodensitométrie pour le diagnostic des kystes cholédociens est controversée. Les kystes de petite taille ou les cholédocèles passent inaperçus à la tomodensitométrie mais sont facilement visibles à la cholangiopancréatographie par résonance magnétique (MRCP). Le scanner peut être meilleur que la MRCP dans la période postopératoire pour détecter l’emplacement de l’anastomose biliaire-entérique et pour définir toute sténose de celle-ci.
La cholangiopancréatographie par résonance magnétique (MRCP) est supérieure pour détecter et définir les lésions. Aujourd’hui, elle est considérée comme l’étalon-or de l’imagerie des kystes cholédociens ; cependant, elle présente l’inconvénient de ne pas pouvoir détecter précisément toute unification pancréaticobiliaire anormale. Néanmoins, cela ne joue pas un rôle majeur dans la détermination de la prise en charge du patient. En outre, la sensibilité de la MRCP est plus importante chez les adultes que chez les nourrissons.
La cholangiopancréatographie rétrograde endoscopique (ERCP) est le meilleur outil pour connaître l’anatomie biliaire et est donc utile pour le diagnostic des kystes cholédociens.
Le traitement comprend principalement une intervention chirurgicale. Une procédure de drainage interne connue sous le nom de kyste-entérostomie (qui pouvait être soit une duodénectomie du kyste, soit une jéjunostomie du kyste) a été réalisée dans le passé pour le traitement chirurgical des kystes cholédociens ; cependant, diverses complications ont été observées en postopératoire, comme une malignité dans le kyste restant, une pancréatite et une cholangite ; elles ont donc été abandonnées.
À l’heure actuelle, la procédure chirurgicale recommandée est l’excision du kyste puis l’hépaticojéjunostomie ou la cholédochojunostomie de Roux-en-Y. L’excision du kyste permet de réduire l’incidence de la formation de sténoses postopératoires. D’autres alternatives, comme la duodénotomie hépatique, ont été proposées pour que l’anastomose soit accessible à la CPRE en cas de complications postopératoires. Cependant, l’utilisation de la duodénotomie hépatique n’a pas été largement acceptée car elle peut provoquer un reflux biliaire et une cholangite.
Après l’excision du kyste, le sondage et le lavage copieux des canaux intrahépatiques avec du sérum physiologique sont nécessaires pour éliminer complètement la boue et les éventuels calculs du système canalaire. De plus, l’obstruction peut être présente dans le système biliaire proximal qui peut être dilaté. Par conséquent, avant l’excision du kyste, une cholangiographie peropératoire est obligatoire.
Défauts génétiques de l’hyperbilirubinémie conjuguée pédiatrique
Il existe de nombreux syndromes héréditaires liés à l’hyperbilirubinémie et à la cholestase intrahépatique. Beaucoup d’entre eux sont liés à des mutations génétiques, notamment le gène SERPINAI (alpha 1- antitrypsine), JAG1 (à l’origine du syndrome d’Alagille), ATP8B1 (également connu sous le nom de FIC1), ABCB11 (pompe d’exportation des sels biliaires ), MDR3 (ABCB4) et MRP2 (à l’origine du syndrome de Dubin-Johnson).
1) Syndrome d’Alagille
Le syndrome d’Alagille est une maladie génétique autosomique dominante qui peut affecter de nombreuses parties du corps. La mutation se produit dans le bras court du chromosome 20 (20p12). Une mutation sur 20 ou 30 se produit de novo. L’un des organes les plus touchés dans le syndrome d’Alagille est le foie et les voies biliaires. Ces malformations des canaux biliaires provoquent une accumulation de bile dans le foie, c’est-à-dire une cholestase, qui entraîne une cicatrisation du foie altérant ainsi son fonctionnement.
Les signes et symptômes découlant de l’atteinte hépatique dans le syndrome d’Alagille peuvent inclure une jaunisse, des démangeaisons de la peau et des xanthomes. Cependant, le cœur est également touché et le patient peut présenter une sténose pulmonaire. En cas de sténose pulmonaire, le flux sanguin du cœur vers les poumons est altéré. Il peut également y avoir des maladies cardiaques congénitales coexistantes, telles que des malformations septales ventriculaires, une tétralogie de Fallot, etc. Le cerveau, la moelle épinière, les reins et les vaisseaux sanguins peuvent également être affectés.
Le syndrome d’Alagille présente des traits faciaux caractéristiques. Le patient a généralement un visage triangulaire, un front large et proéminent, une large arête nasale, des yeux enfoncés et un petit menton pointu. Le diagnostic est difficile et nécessite au moins 3 des caractéristiques physiques typiques, la preuve d’une obstruction du canal biliaire (cholestase) et une biopsie du foie.
Le traitement dépend de la gravité de la maladie dans le syndrome d’Alagille. Dans le cas d’une maladie légère, l’acide ursodésoxycholique est administré pour faciliter le flux biliaire et des antihistaminiques comme la diphénhydramine sont administrés pour contrôler le prurit. Dans les cas graves, une transplantation hépatique peut être nécessaire. Une supplémentation en vitamines s’est avérée utile, notamment en vitamines A, D, E et K. Une supplémentation en zinc est également bénéfique. Ces patients ont une incapacité à absorber ces vitamines et donc, la supplémentation aide à faciliter la croissance optimale du patient.
2) Syndrome de Dubin-Johnson
Ce syndrome est caractérisé par une hyperbilirubinémie chronique conjuguée isolée, sans preuve d’hémolyse due à un défaut d’élimination de la bilirubine conjuguée dans la bile (cholestase intrahépatique). Elle est causée par une mutation du gène MRP2. C’est une affection bénigne qui ne nécessite pas de traitement spécifique.
Syndrome de Rotor
C’est une maladie rare caractérisée par une hyperbilirubinémie chronique conjuguée et non conjuguée sans hémolyse. L’héritage génétique n’est pas encore connu. Elle se produit en raison d’un défaut de stockage de la bilirubine conjuguée dans le foie, qui fuit dans le plasma, provoquant une hyperbilirubinémie.
Etudiez pour la faculté de médecine et les conseils avec Lecturio.
- USMLE Step 1
- USMLE Step 2
- COMLEX Niveau 1
- COMLEX Niveau 2
- ENARMER
- NEET
.