
Bild: „Baby in der Neugeborenen-Intensivstation“ von Jacoplane (na ja, jedenfalls seine Eltern) – Eigenes Werk. Lizenz: CC BY-SA 3.0
- Einführung
- Etiologie der pädiatrischen konjugierten Hyperbilirubinämie
- Diagnose der pädiatrischen konjugierten Hyperbilirubinämie
- Anamnese
- Untersuchung
- Laboruntersuchungen bei pädiatrischer konjugierter Hyperbilirubinämie
- Cholestatische Ursachen der konjugierten Hyperbilirubinämie bei Kindern
- Biliäre Atresie
- Choledochuszysten
- Genetische Defekte der pädiatrischen konjugierten Hyperbilirubinämie
- 1) Alagille-Syndrom
- 2) Dubin-Johnson-Syndrom
- Rotor-Syndrom
Einführung
Bilirubin ist ein Pigment (Tetrapyrrol), das im normalen Stoffwechselweg aus der Häm-Gruppe gebildet wird. Es ist also ein Abfallprodukt des Abbaus reifer roter Blutkörperchen. Entsprechend seinem Stoffwechsel kann es in konjugiertes (korreliert mit direktem Bilirubin) und unkonjugiertes Bilirubin (korreliert mit indirektem Bilirubin) unterteilt werden.
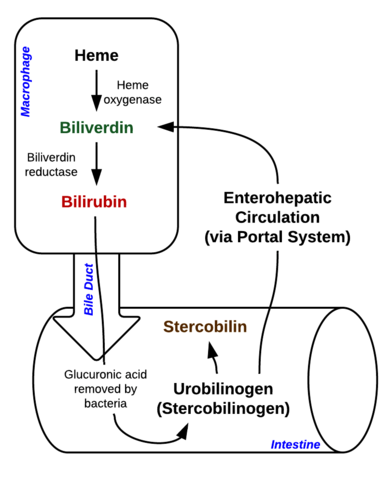
Bild: „Breakdown of Heme in macrophages and intestine“ von Johndheathcote. Lizenz: CC BY-SA 3.0
Hyperbilirubinämie kann definiert werden als eine erhöhte Bilirubinkonzentration im Blut, die sich in Form von Gelbsucht (auch Ikterus genannt) äußert. Gelbsucht tritt auf, wenn sich die gelben Bilirubinpigmente in der Haut, der Sklera, den Schleimhäuten und anderen Geweben ablagern und zu einer gelblichen Verfärbung führen.
Der Normalbereich für Bilirubin liegt bei:
- Gesamtbilirubin im Serum zwischen 0,2 und 1,2 mg/dL.
- Direktes Bilirubin im Serum zwischen 0.1 und 0,4 mg/dL.
Eine Hyperbilirubinämie liegt vor, wenn:
- Direktes Bilirubin im Serum ≥ 1 mg/dL ist (wenn das Gesamtbilirubin < 5mg/dL ist).
- konjugiertes Bilirubin im Serum > 20% des Gesamtbilirubins ist (wenn das Gesamtbilirubin > 5mg/dL ist).
Etiologie der pädiatrischen konjugierten Hyperbilirubinämie
Bei manchen Neugeborenen, insbesondere bei Frühgeborenen, kann ein leichter Anstieg des Bilirubins als normal angesehen werden; er muss jedoch immer untersucht werden, da ein hoher Bilirubinspiegel auf Kosten des konjugierten Bilirubins niemals als normal angesehen werden kann.
Es gibt viele Krankheiten, die eine Hyperbilirubinämie verursachen können, aber sie können grob in zwei Gruppen eingeteilt werden: extrahepatische Störungen (prähepatische Ursachen wie Hämolyse aufgrund intrinsischer Defekte der roten Blutkörperchen oder extrinsischer Ursachen, die zu einer Ruptur der roten Blutkörperchen führen, oder posthepatische Ursachen einschließlich Obstruktion der Gallenwege) und intrahepatische Störungen (Lebererkrankungen).
Es gibt mehrere Ursachen für eine konjugierte Hyperbilirubinämie bei Kindern:
- Infektionen: Virale (Hepatitis A-E, Herpes, Adenovirus, Cytomegalovirus, HIV); bakterielle (Sepsis, Harnwegsinfektionen), Röteln, Tuberkulose, etc.
- Erkrankungen, die zu einer Obstruktion des Gallengangs führen, wie Gallengangsatresie, Perforation des Gallengangs, Cholelithiasis usw.
- Erkrankungen des endokrinen Systems: Hypothyreose, septooptische Dysplasie usw.
- Genetische/metabolische Erkrankungen: Mukoviszidose, Alagille-Syndrom, Alpha-1-Antitrypsin usw.
- Speicherkrankheiten: Gaucher-Syndrom, Glykogenspeicherkrankheiten, Nieman-Pick usw.
- Exposition gegenüber einer Vielzahl von Medikamenten, einschließlich Ceftriaxon, Methotrexat, Erythromycin, Tetracyclin, usw.
Diagnose der pädiatrischen konjugierten Hyperbilirubinämie
Anamnese
Um eine Hyperbilirubinämie bei Kindern zu diagnostizieren, ist es sehr wichtig, eine gute klinische Anamnese zu erheben, einschließlich der pathologischen Vorgeschichte der Eltern und der nahen Verwandten, das mögliche Vorhandensein von Blutsverwandtschaft zwischen den Eltern, mögliche Komplikationen während der Schwangerschaft und der Geburt, mögliche Exposition gegenüber einer Infektionskrankheit vor oder nach der Geburt (wie Hepatitis, Malaria, Zytomegalievirus und Leptospirose) und andere.
Bild: „Neugeborenes Kind, das sich einer Phototherapie zur Behandlung der Neugeborenengelbsucht unterzieht“ von Martin Pot. Lizenz: CC BY 3.0
Untersuchung
Die körperliche Untersuchung ist sehr wichtig für die Diagnose der Hyperbilirubinämie. Das wichtigste Zeichen ist die Gelbsucht oder gelbliche Verfärbung der Sklera, der Schleimhäute und der Haut. Der Urin des Babys färbt sich dunkel, und je nach Ursache kann der Stuhl weiß sein.
Es ist auch äußerst wichtig, eine neurologische Untersuchung des Babys durchzuführen (Reflexe und Reaktion auf äußere Reize), um die Erkrankung des zentralen Nervensystems festzustellen (die als Kernikterus oder Bilirubinenzephalopathie bezeichnet wird). Dies deutet auf das Vorliegen einer schweren Hyperbilirubinämie hin.
Weitere wichtige Parameter, die untersucht werden müssen, sind Wachstumsparameter, Vitalzeichen, Herz- und Atemgeräusche, Bauchuntersuchung (um das abnorme Wachstum von Milz oder Leber festzustellen).
Laboruntersuchungen bei pädiatrischer konjugierter Hyperbilirubinämie
Die erforderlichen Laboruntersuchungen sind:
- Komplettes Blutbild, das für das Screening der Hämolyse erforderlich ist.
- Die Serum-Aminotransferasen werden bestimmt, um die Funktion der Leber zu untersuchen. Dazu gehören Aspartat-Aminotransferase und Alanin-Aminotransferase.
- Serologisches Screening auf Viren.
- Alkalische Phosphatase, da ihr Anstieg auf eine Obstruktion der Gallenwege hinweisen kann.
- Gamma-Glutamyl-Transpeptidase, deren Spiegel dazu beitragen kann, eine hepatische Quelle der erhöhten ALP von anderen Ursachen zu unterscheiden.
- Fraktioniertes Bilirubin.
In anderen Worten, Leberfunktionstests (LFTs oder LFs) sind für die Diagnose notwendig und umfassen Prothrombinzeit (PT/INR), aktivierte partielle Thromboplastinzeit (aPTT), Albumin, Bilirubin (direkt und indirekt) und Lebertransaminasen (AST und ALT). Biopsie und bildgebende Untersuchungen sind in der Regel jenen Fällen vorbehalten, bei denen die Diagnose unklar ist oder obstruktive Pathologien ausgeschlossen werden können.
Cholestatische Ursachen der konjugierten Hyperbilirubinämie bei Kindern
Biliäre Atresie
Die biliäre Atresie ist eine weitere sehr häufige Ursache der konjugierten Hyperbilirubinämie. Der Patient kann eine vollständige oder teilweise Obstruktion des extrahepatischen Gallenbaums an verschiedenen Stellen haben; am häufigsten sind jedoch der Ductus hepaticus und der Ductus bileus betroffen. Eine partielle Obstruktion wird in der Regel durch eine Fibro-Entzündung entlang des Gallengangs verursacht.
Diese Fibro-Entzündung ist auf eine perinatale Erkrankung zurückzuführen, in der Regel eine Virusinfektion, die die Schleimhaut des Gallengangs befällt. Wenn die Infektion fortschreitet, reagiert das Immunsystem und es kommt zu einer Epithelverdickung in der Schleimhaut, die zu einer Obstruktion des Ganges führt. Mit fortschreitender Zeit entwickelt die wuchernde Schleimhaut Sklerose und Fibrose, was im ersten Lebensmonat zu einer totalen Ductusatresie und ihren nachfolgenden Komplikationen führt.
Bei der Diagnose einer Gallengangsatresie ist es ebenfalls sehr wichtig, eine vollständige klinische Anamnese zu erheben, eine gute körperliche Untersuchung und Labortests durchzuführen, wie bereits erwähnt; in diesem Fall können jedoch bildgebende Verfahren (wie Ultraschall, hepatobiliäre Untersuchung (HIDA) oder sogar ein intraoperatives Cholangiogramm) für die Diagnose der Krankheit sehr nützlich sein.
Keine primäre medikamentöse Behandlung ist bei der Korrektur einer Gallengangsatresie wirksam; daher ist, sobald die Diagnose bestätigt ist, ein chirurgischer Eingriff erforderlich, und in vielen Fällen kann die Operation selbst diagnostisch sein. Sie kann in Form eines intraoperativen Cholangiogramms oder einer Kasai-Portoenterostomie durchgeführt werden. Lebertransplantationen sind schweren Fällen vorbehalten.
Choledochuszysten
Schließlich können auch Choledochuszysten, besser bekannt als angeborene Gallengangserweiterung, eine konjugierte Hyperbilirubinämie verursachen. Die häufigste angeborene Anomalie bei Choledochuszysten ist die Erweiterung des extrahepatischen Gallengangs (1 von 100.000-150.000 Neugeborenen). Die Symptome von Choledochuszysten können in jedem Alter auftreten; typischerweise treten sie jedoch bei Säuglingen und Kindern mit obstruktiver Gelbsucht und Bauchschmerzen auf. Sie treten bevorzugt bei Frauen auf, wobei das Verhältnis von Frauen zu Männern etwa 3-4:1 beträgt. Sie sind bei bestimmten asiatischen Rassen häufiger anzutreffen.
Typen von Choledochuszysten: Ursprünglich wurden drei Typen von Choledochuszysten beschrieben, aber Todani und Mitarbeiter haben diese in fünf Typen unterteilt, die im Folgenden beschrieben werden:
- Typ I: Es liegt eine Dilatation des Hauptgallengangs vor. Sie kann zystisch, fokal oder fusiform sein (in 90-95 % der Fälle).
- Typ II: Hier wird ein Divertikel des extrahepatischen Gallengangs gesehen.
- Typ III: Gekennzeichnet durch Choledochozelen.
- Typ IV: Es gibt zwei Typen, Typ IV-A und Typ IV-B. Typ IV-A ist der zweithäufigste Typ und wird als intrahepatische und extrahepatische Dilatation des Gallenganges definiert. Beim Typ IV-B handelt es sich um die seltene Fehlbildung multipler extrahepatischer Zysten.
- Typ V: Dieser Typ wird auch als Caroli-Krankheit bezeichnet, wenn er mit Leberfibrose einhergeht. Typ V umfasst einzelne oder mehrere intrahepatische Zysten.
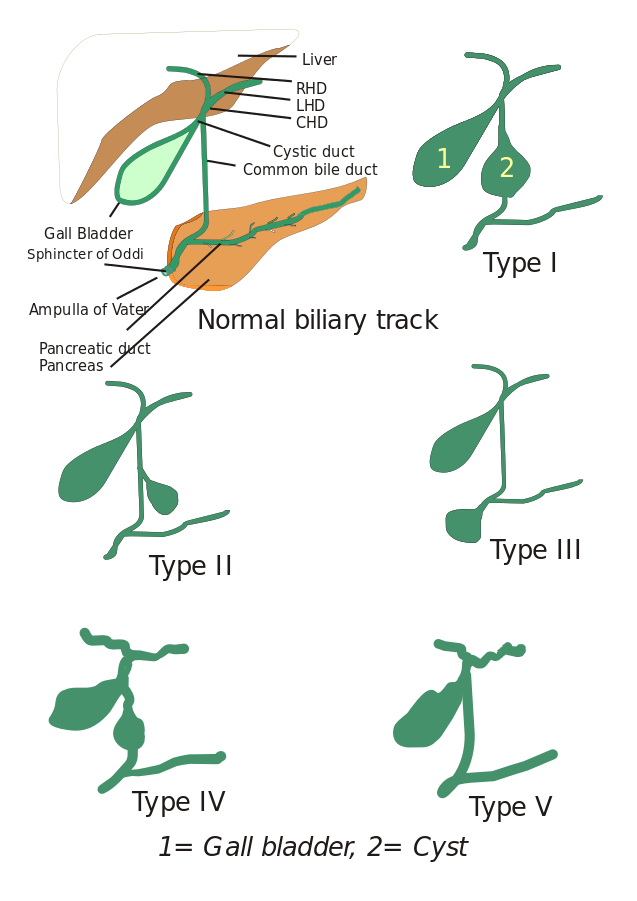
Bild: „5 Typen von Choledochuszysten. Typ I: Dilatation des extrahepatischen Gallengangs; Typ II: Zyste aus dem Hauptgallengang (CBD); Typ III: Choledochozele oder Dilatation des distalen Teils des CBD, Typ IV: Dilatation sowohl des extrahepatischen als auch des intrahepatischen Gangs; Typ V: Morbus Caroli, Dilatation nur des Ductus intrahepaticus. CHD: Gemeinsamer Ductus hepaticus, LHD: Linker Ductus hepaticus und RHD: Rechter Ductus hepaticus.“ Von I, Drriad. Lizenz: CC BY-SA 3.0
Sie tritt häufig bei Säuglingen im Alter von 1 bis 3 Monaten auf. Klinisch zeigen sich Fieber, Schmerzen im rechten oberen Quadranten des Abdomens, acholischer oder blasser Stuhl und Hepatomegalie, und dieses Bild ähnelt einer biliären Atresie. Im Falle einer pränatalen Diagnose einer Choledochuszyste zeigt sich die Gelbsucht erst 1 bis 3 Wochen nach der Geburt. Wenn die Choledochuszyste erst später auftritt, wird sie erst nach dem zweiten Lebensjahr klinisch sichtbar und kann sich mit einer klassischen Trias aus Bauchschmerzen, tastbarer abdominaler Masse und Gelbsucht präsentieren, von denen zwei bei fast 85 % der Kinder zum Zeitpunkt der Präsentation zu finden sind. Weitere häufige Merkmale sind Cholangitis, Pankreatitis und biliäre Peritonitis aufgrund der Ruptur der Zyste.
Für eine endgültige Diagnose sind Laboruntersuchungen sowie bildgebende Verfahren erforderlich. Zu den Laboruntersuchungen gehören die üblichen Leberfunktionstests, das große Blutbild und Gerinnungsprofile. Zu den bildgebenden Untersuchungen gehören u. a. Ultraschall, Computertomographie, Magnetresonanztomographie, Magnetresonanz-Cholangiopankreatographie und endoskopische retrograde Cholangiopankreatographie.
Die Ultraschalluntersuchung hilft bei der Diagnose von Choledochuszysten und hat eine sehr hohe Spezifität von 97 % bei Kindern, aber eine geringe diagnostische Genauigkeit vorgeburtlich; daher wird sie als primäre diagnostische Bildgebungsmethode bei Neugeborenengelbsucht eingesetzt, die mehr als 2 Wochen nach der Geburt besteht. Sie ist auch nützlich bei der Unterscheidung zwischen Choledochuszysten und biliärer Atresie.
Die Verwendung von Computertomographien zur Diagnose von Choledochuszysten ist umstritten. Kleine Zysten oder Choledochozele werden auf CT-Scans übersehen, sind aber auf der Magnetresonanz-Cholangiopankreatographie (MRCP) leicht zu erkennen. In der postoperativen Phase kann die CT besser als die MRCP geeignet sein, um die Lage der biliären-enterischen Anastomose zu erkennen und deren Stenose zu definieren.
Die Magnetresonanz-Cholangiopankreatographie (MRCP) ist besser geeignet, Läsionen zu erkennen und zu definieren. Sie gilt heute als Goldstandard in der Bildgebung von Choledochuszysten, hat jedoch den Nachteil, dass sie nicht in der Lage ist, eine anomale pankreatisch-biliäre Vereinigung genau zu erkennen. Dies spielt jedoch bei der Festlegung des Patientenmanagements keine große Rolle. Außerdem ist die Sensitivität der MRCP bei Erwachsenen höher als bei Kindern.
Die endoskopische retrograde Cholangiopankreatographie (ERCP) ist das beste Instrument zur Kenntnis der Gallengangsanatomie und somit hilfreich bei der Diagnose von Choledochuszysten.
Die Behandlung umfasst in erster Linie einen chirurgischen Eingriff. In der Vergangenheit wurde zur chirurgischen Behandlung von Choledochuszysten ein internes Drainageverfahren durchgeführt, das als Zysten-Enterostomie (entweder als Zysten-Duodenektomie oder als Zysten-Jejunostomie) bekannt ist; allerdings traten postoperativ verschiedene Komplikationen auf, wie z. B. Malignität in der verbleibenden Zyste, Pankreatitis und Cholangitis; daher wurden sie aufgegeben.
Das derzeit empfohlene chirurgische Verfahren ist die Zystenexzision mit anschließender Roux-en-Y-Hepaticojejunostomie oder Choledochojejunostomie. Durch die Zystenexzision wird die Inzidenz der Strikturbildung nach der Operation verringert. Andere Alternativen, z. B. die hepatische Duodenotomie, wurden vorgeschlagen, damit die Anastomose im Falle postoperativer Komplikationen für die ERCP zugänglich ist. Die hepatische Duodenotomie hat sich jedoch nicht durchgesetzt, da sie zu einem biliären Reflux und einer Cholangitis führen kann.
Nach der Zystenexzision ist eine Sondierung und ausgiebige Spülung der intrahepatischen Gänge mit Kochsalzlösung erforderlich, um Schlamm und mögliche Steine vollständig aus dem Gangsystem zu entfernen. Darüber hinaus kann die Obstruktion im proximalen Gallengangssystem vorhanden sein, das erweitert werden kann. Daher ist vor der Exzision der Zyste eine intraoperative Cholangiographie obligatorisch.
Genetische Defekte der pädiatrischen konjugierten Hyperbilirubinämie
Es gibt viele vererbte Syndrome im Zusammenhang mit Hyperbilirubinämie und intrahepatischer Cholestase. Viele von ihnen hängen mit genetischen Mutationen zusammen, darunter die Gene SERPINAI (Alpha-1-Antitrypsin), JAG1 (verursacht das Alagille-Syndrom), ATP8B1 (auch bekannt als FIC1), ABCB11 (Gallensalzexportpumpe ), MDR3 (ABCB4) und MRP2 (verursacht das Dubin-Johnson-Syndrom).
1) Alagille-Syndrom
Das Alagille-Syndrom ist eine autosomal dominante genetische Störung, die viele Teile des Körpers betreffen kann. Die Mutation tritt auf dem kurzen Arm von Chromosom 20 (20p12) auf. Eine von 20 bis 30 Mutationen tritt de novo auf. Eines der am häufigsten betroffenen Organe beim Alagille-Syndrom sind die Leber und die Gallengänge. Diese Fehlbildungen der Gallengänge führen zu einer Ansammlung von Gallenflüssigkeit in der Leber, d. h. zu Cholestase, die eine Vernarbung der Leber verursacht und dadurch ihre Funktion beeinträchtigt.
Zu den Anzeichen und Symptomen, die durch die Leberschädigung beim Alagille-Syndrom entstehen, gehören Gelbsucht, juckende Haut und Xanthome. Aber auch das Herz ist betroffen und der Patient kann eine Pulmonalstenose haben. Bei der Pulmonalstenose ist der Blutfluss vom Herzen in die Lunge beeinträchtigt. Es können auch gleichzeitig angeborene Herzkrankheiten wie Ventrikelseptumdefekte, Fallot-Tetralogie usw. vorliegen. Auch das Gehirn, das Rückenmark, die Nieren und die Blutgefäße können betroffen sein.
Das Lagille-Syndrom hat charakteristische Gesichtszüge. Der Patient hat in der Regel ein dreieckiges Gesicht, eine breite und markante Stirn, einen breiten Nasenrücken, tief liegende Augen und ein kleines, spitzes Kinn. Die Diagnose ist schwierig und erfordert mindestens 3 der typischen körperlichen Merkmale, den Nachweis einer Gallengangsobstruktion (Cholestase) und eine Leberbiopsie.
Die Behandlung des Alagille-Syndroms richtet sich nach dem Schweregrad der Erkrankung. Im Falle einer leichten Erkrankung wird Ursodeoxycholsäure verabreicht, um den Gallenfluss zu erleichtern, und Antihistaminika wie Diphenhydramin, um den Juckreiz zu kontrollieren. In schweren Fällen kann eine Lebertransplantation erforderlich sein. Eine Vitaminergänzung hat sich als hilfreich erwiesen, insbesondere mit den Vitaminen A, D, E und K. Eine Zinkergänzung ist ebenfalls von Vorteil. Diese Patienten sind nicht in der Lage, diese Vitamine zu absorbieren, so dass eine Supplementierung ein optimales Wachstum der Patienten fördert.
2) Dubin-Johnson-Syndrom
Dieses Syndrom ist gekennzeichnet durch eine isolierte konjugierte chronische Hyperbilirubinämie ohne Anzeichen einer Hämolyse aufgrund eines Defekts bei der Ausscheidung von konjugiertem Bilirubin in der Galle (intrahepatische Cholestase). Sie wird durch eine Mutation im MRP2-Gen verursacht. Es handelt sich um eine gutartige Erkrankung, die keine spezifische Behandlung erfordert.
Rotor-Syndrom
Es handelt sich um eine seltene Störung, die durch chronische konjugierte und unkonjugierte Hyperbilirubinämie ohne Hämolyse gekennzeichnet ist. Der genetische Erbgang ist noch nicht bekannt. Sie tritt aufgrund eines Defekts der Speicherung von konjugiertem Bilirubin in der Leber auf, das in das Plasma austritt und eine Hyperbilirubinämie verursacht.
Mit Lecturio für das Medizinstudium und die Prüfungen lernen.
- USMLE Step 1
- USMLE Step 2
- COMLEX Level 1
- COMLEX Level 2
- ENARM
- NEET