
Obrázek: „Dítě na novorozenecké jednotce intenzivní péče.“ Autor: Jacoplane (tedy jeho rodiče) – vlastní tvorba. Licence: CC BY-SA 3.0
- Úvod
- Etiologie dětské konjugované hyperbilirubinémie
- Diagnostika dětské konjugované hyperbilirubinémie
- Historie
- Vyšetření
- Laboratorní vyšetření dětské konjugované hyperbilirubinémie
- Cholestatické příčiny dětské konjugované hyperbilirubinémie
- Biliární atrézie
- Cysty choledochu
- Genetické vady dětské konjugované hyperbilirubinémie
- 1) Alagillův syndrom
- 2) Dubin-Johnsonův syndrom
- Rotorův syndrom
Úvod
Bilirubin je pigment (tetrapyrrol), který vzniká normální katabolickou cestou z hemové skupiny. Jedná se tedy o odpadní produkt destrukce zralých červených krvinek. Podle jeho metabolismu jej lze klasifikovat jako konjugovaný (koreluje s přímým bilirubinem) a nekonjugovaný bilirubin (koreluje s nepřímým bilirubinem).
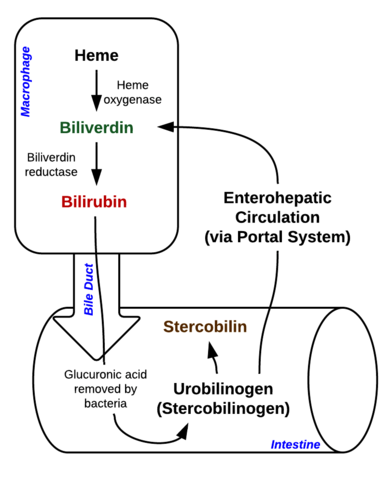
Obrázek: Johndheathcote: „Breakdown of Heme in macrophages and intestine“. Licence: CC BY-SA 3.0
Hyperbilirubinémii lze definovat jako zvýšenou koncentraci bilirubinu v krvi, která se projevuje žloutenkou (nazývanou také ikterus). Žloutenka se objevuje, když se žluté pigmenty bilirubinu ukládají v kůži, sklérách, sliznicích a dalších tkáních, což vede ke žlutavému zbarvení.
Normální rozmezí bilirubinu je:
- Celkový bilirubin v séru mezi 0,2 a 1,2 mg/dl.
- Přímý bilirubin v séru mezi 0,2 a 1,2 mg/dl.1 až 0,4 mg/dl.
Za hyperbilirubinémii se považuje, když:
- Přímý bilirubin v séru je ≥ 1 mg/dl (pokud je celkový bilirubin < 5mg/dl).
- Konjugovaný bilirubin v séru je > 20% celkového bilirubinu (pokud je celkový bilirubin > 5mg/dl).
Etiologie dětské konjugované hyperbilirubinémie
U některých novorozenců, zejména nedonošených, lze mírné zvýšení bilirubinu považovat za normální; vždy je však třeba jej vyšetřit, protože vysokou hladinu bilirubinu na úkor konjugovaného bilirubinu nelze nikdy považovat za normální.
Existuje mnoho onemocnění, která mohou způsobit hyperbilirubinémii, ale lze je obecně rozdělit do dvou skupin: extrahepatální poruchy (prehepatální příčiny, jako je hemolýza způsobená vnitřními defekty červených krvinek nebo vnější příčiny vedoucí k prasknutí červených krvinek, nebo posthepatální příčiny včetně obstrukce žlučových cest) a intrahepatální poruchy (onemocnění jater).
U dětí existuje několik stavů konjugované hyperbilirubinémie :
- Infekce: Virové (hepatitida A-E, herpes, adenovirus, cytomegalovirus, HIV); bakteriální (sepse, infekce močových cest), zarděnky, tuberkulóza atd.
- Nemoci, které způsobují obstrukci žlučových cest, jako je atrézie žlučových cest, perforace žlučových cest, cholelitiáza atd
- Nemoci endokrinního systému: hypotyreóza, septooptická dysplazie atd.
- Genetické/metabolické: cystická fibróza, Alagillův syndrom, alfa-1-antitrypsin atd.
- Poruchy skladování:
- Expozice mnoha lékům včetně ceftriaxonu, metotrexátu, erytromycinu, tetracyklinu atd.
Diagnostika dětské konjugované hyperbilirubinémie
Historie
Pro stanovení diagnózy hyperbilirubinémie u dětí je velmi důležité odebrat dobrou klinickou anamnézu, včetně patologických anamnéz rodičů a blízkých příbuzných, možnou existenci příbuzenství mezi rodiči, možné komplikace během těhotenství a porodu, možné vystavení infekčnímu onemocnění před narozením nebo po něm (např. hepatitida, malárie, cytomegalovirus a leptospiróza) a další.
Obr: „Novorozenec podstupující fototerapii k léčbě novorozenecké žloutenky“, autor: Martin Pot. Licence: CC BY 3.0
Vyšetření
Fyzikální vyšetření je při diagnostice hyperbilirubinémie velmi důležité. Hlavním příznakem je žloutenka nebo nažloutlé zbarvení skléry, sliznic a kůže. Moč dítěte tmavne a v závislosti na příčině může být bílá i stolice.
Je také nesmírně důležité provést neurologické vyšetření dítěte (reflexy a reakce na vnější podněty), aby bylo možné určit postižení centrálního nervového systému (které je známé jako kernicterus nebo bilirubinová encefalopatie). To ukazuje na přítomnost těžké hyperbilirubinémie.
Dalšími důležitými parametry, které je třeba vyšetřit, jsou růstové parametry, životní funkce, srdeční a dechové šelesty, vyšetření břicha (za účelem zjištění abnormálního růstu sleziny nebo jater).
Laboratorní vyšetření dětské konjugované hyperbilirubinémie
Potřebná laboratorní vyšetření jsou:
- Kompletní krevní obraz, který je nezbytný pro screening hemolýzy.
- Hladina aminotransferáz v séru se provádí za účelem vyšetření funkce jater. Zahrnují aspartátaminotransferázu a alaninaminotransferázu .
- Serologický screening na viry.
- Alkalická fosfatáza, protože její zvýšení může ukazovat na obstrukci žlučových cest.
- Gamma-glutamyltranspeptidáza, jejíž hladiny mohou pomoci odlišit jaterní zdroj zvýšené ALP od jiných příčin.
- Frakcionovaný bilirubin.
Jinak jsou pro stanovení diagnózy nezbytné jaterní funkční testy (LFT nebo LF), které zahrnují protrombinový čas (PT/INR), aktivovaný parciální tromboplastinový čas (aPTT), albumin, bilirubin (přímý a nepřímý) a jaterní transaminázy (AST a ALT). Biopsie a zobrazovací vyšetření jsou obvykle vyhrazeny pro případy s nejasnou diagnózou nebo vyřazení obstrukčních patologií.
Cholestatické příčiny dětské konjugované hyperbilirubinémie
Biliární atrézie
Biliární atrézie je další velmi častou příčinou konjugované hyperbilirubinémie. Pacient může mít úplnou nebo částečnou obstrukci extrahepatálního žlučového stromu na různých místech; nejčastěji však bývají postiženy jaterní vývod a společný žlučovod. Částečná obstrukce je obvykle způsobena fibro-zánětem podél žlučovodu.
Tento fibro-zánět je způsoben perinatálním onemocněním, obvykle virovou infekcí, která postihuje sliznici žlučových cest. Při progresi infekce reaguje imunologický systém a dochází ke ztluštění epitelu sliznice, což vede k obstrukci vývodu. S postupujícím časem dochází u prorůstající sliznice ke skleróze a fibróze, což způsobuje úplnou atrézii vývodu během prvního měsíce života a její následné komplikace.
Při diagnostice atrézie žlučových cest je rovněž velmi důležité odebrat kompletní klinickou anamnézu, provést kvalitní fyzikální vyšetření a laboratorní testy, jak bylo uvedeno výše; v tomto případě však mohou být pro diagnostiku onemocnění velmi užitečné zobrazovací techniky (jako ultrazvuk, hepatobiliární scan (HIDA) nebo dokonce intraoperační cholangiogram).
Žádná primární medikamentózní léčba není při korekci biliární atrézie účinná, proto je po potvrzení diagnózy nutný chirurgický zákrok a v mnoha případech může být samotná operace diagnostická. Může být proveden jako intraoperační cholangiogram nebo Kasaiova portoenterostomie. Transplantace jater je vyhrazena pro těžké případy.
Cysty choledochu
Nakonec, cysty choledochu, známější jako vrozená dilatace žlučových cest, mohou také způsobit konjugovanou hyperbilirubinémii. Nejčastější vrozenou abnormalitou choledochálních cyst je rozšíření extrahepatálního žlučovodu (1 z každých 100 000-150 000 novorozenců). Příznaky choledochálních cyst se mohou projevit v jakémkoli věku, charakteristická je však obstrukční žloutenka spolu s bolestmi břicha u kojenců a dětí. Mají predilekci u žen, poměr žen a mužů je přibližně 3-4:1. Častěji se vyskytují u některých asijských ras.
Typy choledochálních cyst: Původně byly popsány tři typy choledochálních cyst, ale Todani a spolupracovníci je dále rozdělili do pěti typů, které jsou popsány níže:
- Typ I: Jedná se o dilataci společného žlučovodu. Může být cystická, fokální nebo fusiformní (vyskytuje se v 90-95 % případů).
- Typ II: Je zde patrný divertikl extrahepatálního žlučovodu.
- Typ III: Vyznačuje se choledochokélou.
- Typ IV: Rozlišují se dva typy, typ IV-A a typ IV-B.
- Typ IV: Vyskytuje se ve dvou případech. Typ IV-A je druhým nejčastějším typem a je definován jako intrahepatální i extrahepatální dilatace žlučového stromu. Typ IV-B zahrnuje vzácnou malformaci mnohočetných extrahepatálních cyst.
- Typ V: Tento typ je také znám jako Caroliho nemoc, pokud je spojen s jaterní fibrózou. Typ V zahrnuje jednu nebo vícečetné intrahepatální cysty.
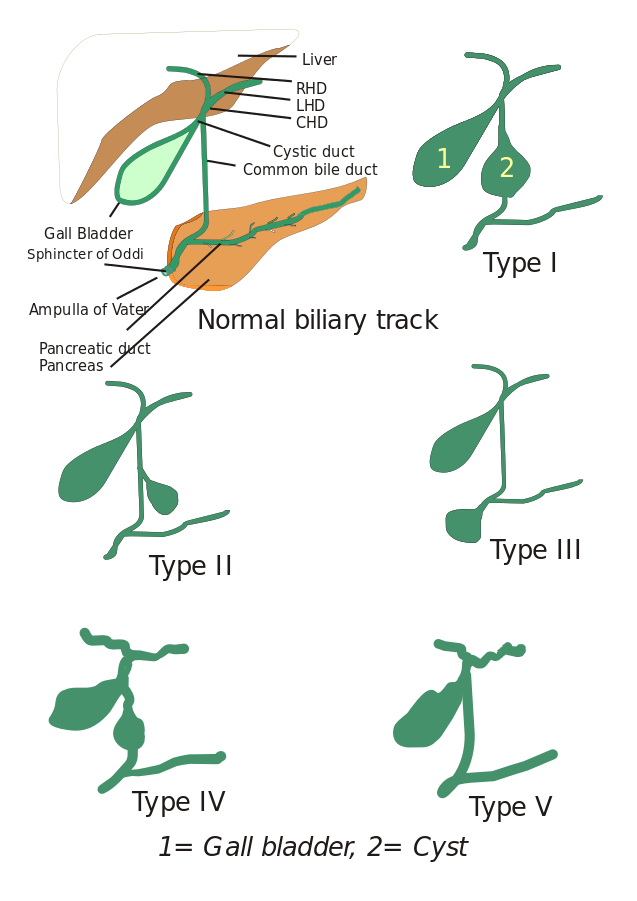
Obrázek: Typ V zahrnuje jednu nebo vícečetné intrahepatální cysty: „5 typů choledochálních cyst. Typ I: Typ II: cysta ze společného žlučovodu (CBD); Typ III: choledochokéla nebo dilatace distální části CBD, typ IV: dilatace extrahepatálního i intrahepatálního žlučovodu; Typ V: Caroliho choroba, dilatace pouze intrahepatálního vývodu. CHD: společný jaterní vývod, LHD: levý jaterní vývod a RHD: RHD: pravý jaterní vývod.“ Podle I, Drriad. Licence: Běžně se vyskytuje u kojenců ve věku od 1 do 3 měsíců: CC BY-SA 3.0
. Klinicky se projevují horečkou, bolestí v pravém horním kvadrantu břicha, acholickou nebo bledou stolicí a hepatomegalií a tento obraz je podobný biliární atrézii. V případě prenatální diagnózy choledochální cysty se žloutenka projeví až 1 až 3 týdny po narození. Pokud se choledochální cysta objeví později, klinicky se projeví až po 2 letech věku a může se projevit klasickou triádou bolestí břicha, hmatné břišní masy a žloutenky; z toho dvě se vyskytují u téměř 85 % dětí v době prezentace. Dalšími častými příznaky jsou cholangitida, pankreatitida a biliární peritonitida v důsledku ruptury cysty.
K definitivní diagnóze jsou nezbytná laboratorní vyšetření a také zobrazovací vyšetření. Laboratorní vyšetření zahrnují konvenční testy jaterních funkcí, krevní obraz a koagulační profily. Zobrazovací vyšetření zahrnují mimo jiné ultrazvukové vyšetření, počítačovou tomografii, magnetickou rezonanci, magnetickou rezonanční cholangiopankreatografii a endoskopickou retrográdní cholangiopankreatografii.
Ultrazvukové vyšetření pomáhá při diagnostice choledochálních cyst a má velmi vysokou specificitu 97 % u dětí, ale nízkou diagnostickou přesnost antenatálně; proto se používá jako primární diagnostická zobrazovací metoda u novorozenecké žloutenky, která přetrvává déle než 2 týdny po porodu. Je také užitečná při odlišení choledochálních cyst a biliární atrézie.
Použití počítačové tomografie pro diagnostiku choledochálních cyst je kontroverzní. Cysty malých rozměrů nebo choledochokéla jsou na CT skenech přehlédnuty, ale jsou snadno viditelné na magnetické rezonanční cholangiopankreatografii (MRCP). CT vyšetření může být v pooperačním období lepší než MRCP při zjišťování polohy biliárně-enterické anastomózy a při definování její případné stenózy.
Magnetická rezonanční cholangiopankreatografie (MRCP) je lepší při zjišťování a definování lézí. V současné době je považována za zlatý standard při zobrazování choledochálních cyst; její nevýhodou však je, že není schopna přesně detekovat případné anomální pankreatobiliární sjednocení. Nicméně to nehraje zásadní roli při určování managementu pacienta. Kromě toho je citlivost MRCP větší u dospělých než u kojenců.
Endoskopická retrográdní cholangiopankreatografie (ERCP) je nejlepším nástrojem pro poznání anatomie žlučových cest, a je tak užitečná při diagnostice choledochálních cyst.
Léčba zahrnuje především chirurgickou intervenci. V minulosti se při chirurgické léčbě choledochálních cyst prováděla vnitřní drenáž známá jako cysto-enterostomie (která může být buď cysto-duodenektomie, nebo cysto-jejunostomie); pooperačně se však vyskytovaly různé komplikace, např. malignita ve zbývající cystě, pankreatitida a cholangitida, a proto se od nich upustilo.
V současné době je doporučovaným chirurgickým postupem excize cysty a následně Roux-en-Y hepatikojejunostomie nebo choledochojejunostomie. Vzhledem k excizi cysty dochází ke snížení výskytu tvorby striktur v pooperačním období. Byly navrženy další alternativy, tj. jaterní duodenotomie, aby byla anastomóza přístupná ERCP v případě pooperačních komplikací. Použití jaterní duodenotomie však nebylo široce přijato, protože může způsobit biliární reflux a cholangitidu.
Po excizi cysty je k úplnému odstranění kalu a případných kamenů z vývodného systému nutné sondování a hojný výplach intrahepatálních vývodů fyziologickým roztokem. Dále může být obstrukce přítomna v proximálním žlučovém systému, který může být dilatován. V důsledku toho je před excizí cysty povinná intraoperační cholangiografie.
Genetické vady dětské konjugované hyperbilirubinémie
Existuje mnoho dědičných syndromů souvisejících s hyperbilirubinémií a intrahepatální cholestázou. Mnoho z nich souvisí s genetickými mutacemi, včetně genu SERPINAI (alfa 1- antitrypsin), JAG1 (způsobuje Alagillův syndrom), ATP8B1 (známý také jako FIC1), ABCB11 (pumpa pro export žlučových solí ), MDR3 (ABCB4) a MRP2 (způsobuje Dubin-Johnsonův syndrom).
1) Alagillův syndrom
Alagillův syndrom je autozomálně dominantní genetické onemocnění, které může postihovat mnoho částí těla. Mutace se vyskytuje v krátkém raménku 20. chromozomu (20p12). Jedna z 20 až 30 mutací se vyskytuje de novo. Jedním z nejvíce postižených orgánů u Alagillova syndromu jsou játra a žlučovody. Tyto malformace žlučových cest způsobují hromadění žluči v játrech, tj. cholestázu, která způsobuje jizvení jater, čímž se mění jejich fungování.
Známky a příznaky vyplývající z poškození jater u Alagillova syndromu mohou zahrnovat žloutenku, svědění kůže a xantomy. Postiženo je však také srdce a pacient může mít pulmonální stenózu. Při pulmonální stenóze dochází k poruše průtoku krve ze srdce do plic. Současně se mohou vyskytovat vrozené srdeční vady, jako jsou defekty komorového septa, Fallotova tetralogie atd. Postižen může být také mozek, mícha, ledviny a cévy.
Alagillův syndrom má charakteristické rysy obličeje. Pacient má obvykle trojúhelníkovitý obličej, široké a výrazné čelo, široký nosní můstek, hluboko posazené oči a malou špičatou bradu. Diagnóza je obtížná a vyžaduje alespoň 3 typické fyzikální znaky, průkaz obstrukce žlučových cest (cholestázy) a jaterní biopsii.
Léčba je u Alagillova syndromu závislá na závažnosti onemocnění. V případě mírného onemocnění se podává kyselina ursodeoxycholová k usnadnění průtoku žlučí a antihistaminika, jako je difenhydramin, ke kontrole pruritu. U těžkých případů může být nutná transplantace jater. Bylo zjištěno, že pomáhá suplementace vitaminy, zejména vitaminy A, D, E a K. Přínosná je také suplementace zinkem. Tito pacienti mají neschopnost vstřebávat tyto vitaminy, a proto suplementace pomáhá při usnadnění optimálního růstu pacienta.
2) Dubin-Johnsonův syndrom
Tento syndrom je charakterizován izolovanou konjugovanou chronickou hyperbilirubinémií bez známek hemolýzy v důsledku defektu v eliminaci konjugovaného bilirubinu ve žluči (intrahepatální cholestáza). Je způsobena mutací v genu MRP2. Jedná se o benigní onemocnění, které nevyžaduje specifickou léčbu.
Rotorův syndrom
Jedná se o vzácnou poruchu charakterizovanou chronickou konjugovanou a nekonjugovanou hyperbilirubinémií bez hemolýzy. Genetická dědičnost není dosud známa. Vzniká v důsledku poruchy ukládání konjugovaného bilirubinu v játrech, který uniká do plazmy a způsobuje hyperbilirubinémii.
Studujte na lékařskou fakultu a komise s Lecturio.
- USMLE Step 1
- USMLE Step 2
- COMLEX Level 1
- COMLEX Level 2.
- ENARM
- NEET