
Lisossomas são organelas esféricas, ligadas por membrana que são geradas pelo aparelho golgi. Elas contêm enzimas hidrolíticas, e por isso funcionam como parte do sistema de reciclagem da célula.
Neste artigo, vamos olhar para a estrutura, síntese e função dos lisossomos, e vamos considerar sua relevância para a prática clínica.
Estrutura
Os lisossomos são organelas amarradas a membrana ácida encontradas dentro das células, geralmente em torno de 1 micrómetro de comprimento. Os lisossomas contêm numerosas enzimas hidrolíticas que catalisam reacções de hidrólise.
A membrana que envolve o lisossoma é vital para assegurar que estas enzimas não vazem para o citoplasma e danifiquem a célula a partir de dentro. A fim de manter o pH ácido do lisossoma, os prótons são ativamente transportados para a organela através da membrana lisossômica.
Síntese
O lisossoma e as enzimas dentro dele são sintetizados separadamente. As proteínas lisossômicas são formadas da mesma forma que qualquer outra proteína. O primeiro passo é a iniciação da produção do mRNA a partir de segmentos de DNA relevantes. Os filamentos de mRNA seguem para o retículo endoplasmático grosso, onde ribossomos constroem as enzimas hidrolíticas.
Importantemente, estas são marcadas com manose-6-fosfato dentro do aparelho de Golgi para direcioná-las ao lisossomo. Como resultado, vesículas contendo estas enzimas brotam do aparelho de Golgi. Duas enzimas são responsáveis pela fixação da etiqueta do manose-6-fosfato: N-acetilglucosamina fosfotransferase e N-acetilglucosamina fosfoglicosidase.
Esta vesícula, agora no citoplasma, liga-se com um endossoma tardio que é outra organela ácida, ligada à membrana. O endossoma tardio tem bombas de prótons dentro da sua membrana que mantêm o seu ambiente interno ácido. O pH baixo causa dissociação da proteína do receptor manose-6-fosfato. Este receptor pode então ser reciclado de volta ao aparelho de Golgi.
O grupo fosfato também é removido do rótulo manose-6-fosfato, para evitar que a proteína inteira volte ao aparelho de Golgi. O endossomo tardio pode eventualmente amadurecer em um lisossomo, após ter recebido as enzimas do aparelho de Golgi.
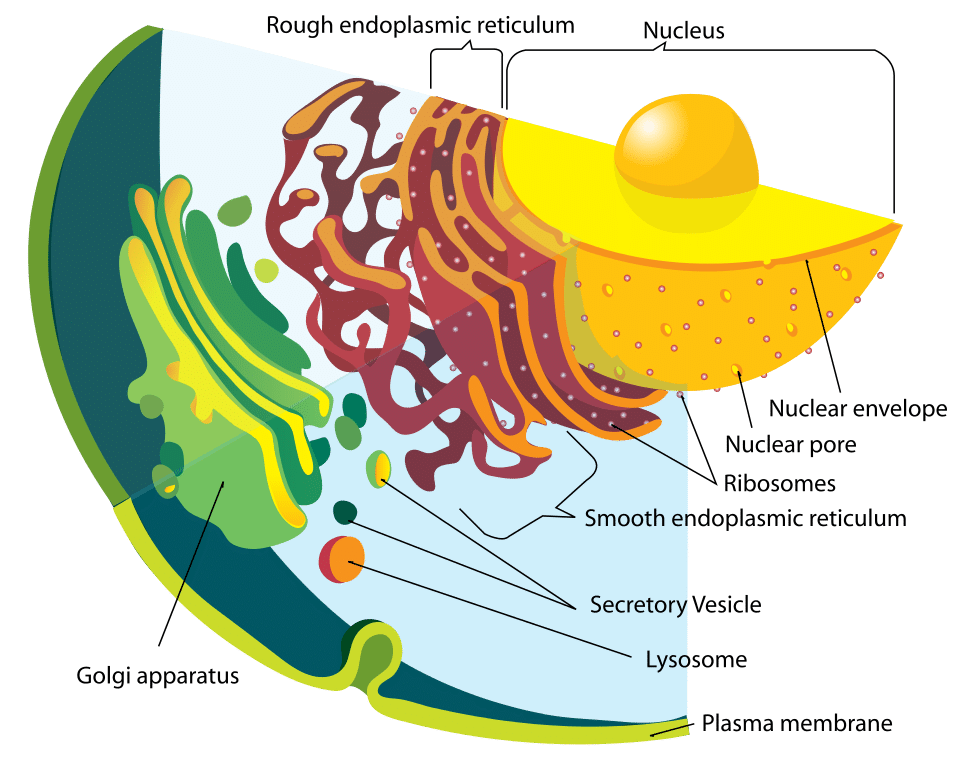 Figura 1 – Diagrama do sistema endomembrana
Figura 1 – Diagrama do sistema endomembranaFunção
As enzimas hidrolíticas contidas no lisossomo permitem que partículas estranhas sejam destruídas. Os lisossomas desempenham um papel importante na fagocitose. Quando os macrófagos fagócitos contêm partículas estranhas, eles contêm-nas dentro de um fagosoma. O fagosoma irá então ligar-se com um lisossoma para formar um fagosoma.
Estas enzimas são críticas nos mecanismos de morte independentes do oxigénio. Os lisossomas também ajudam a defender contra a entrada de patógenos através da endocitose, degradando os patógenos antes que eles alcancem o citoplasma.
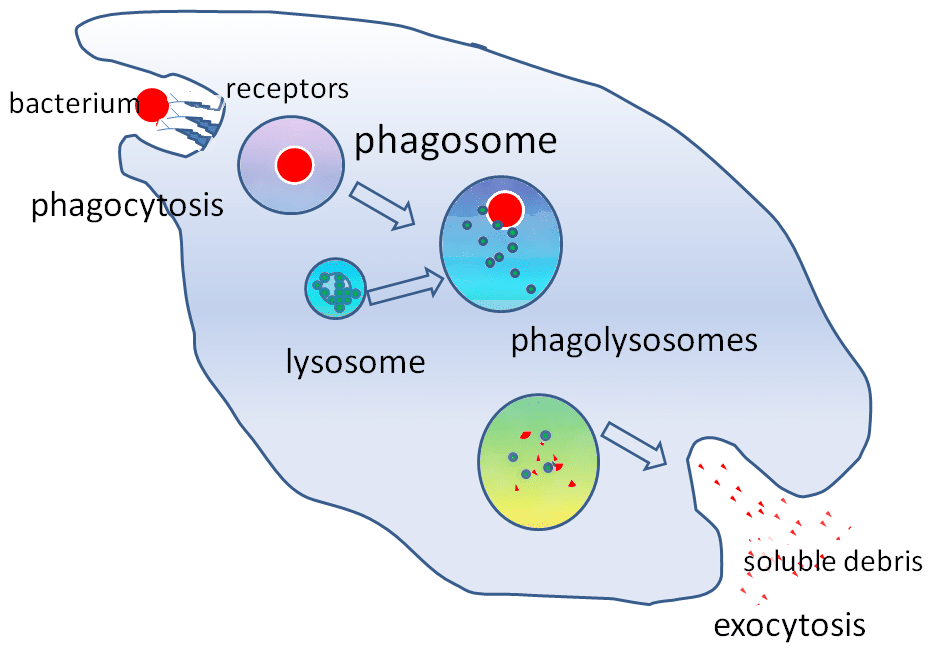 Figura 2 – O papel do lisossoma na fagocitose
Figura 2 – O papel do lisossoma na fagocitosePertinência clínica
Doença das células I
Proveniente de defeitos genéticos na enzima N-acetilglucosamina fosfotransferase. Esta enzima é vital para a adição de manose-6-fosfato às proteínas visadas pelo lisossoma. Isto resulta em enzimas lisossômicas não sendo devidamente visadas. Como resultado, quantidades significativas são encontradas tanto na urina quanto na corrente sanguínea.
Doença de armazenamento lisossômico
Essas são um grupo de condições genéticas que afetam os lisossomos. A condição varia muito em sinais, sintomas e demografia do paciente. Existem várias classificações; a mais comum destas condições é a doença de Gaucher. É causada por uma deficiência da beta-glucocerebrosidase. Esta enzima é necessária para quebrar a glucocerebrosidase. Sem esta enzima, a glucocerebrosidase é capaz de se acumular dentro das células, o que pode danificá-las. Os sintomas incluem hepatoesplenomegalia e anemia.