
Sinais e Sintomas
SMA ligado ao cromossoma 5 (relacionado ao SMN), tipos 0-4
Na atrofia muscular espinhal (SMA) tipos 0 a 4, os sintomas variam em um contínuo de severo a leve com base na quantidade de proteína SMN funcional que existe nas células nervosas chamadas neurônios motores. (“SMN” significa sobrevivência do neurônio motor.) Quanto mais proteína SMN houver, mais tarde começam os sintomas na vida e mais leve o curso da doença. O número de cópias dos genes SMN2, que podem compensar o mau funcionamento dos genes SMN1, determina a gravidade da AME e a idade de início do paciente.
As mães de crianças afectadas por AME tipo 0 podem relatar uma diminuição do movimento fetal no final da gravidez e dar à luz mais cedo. Estes recém-nascidos apresentam fraqueza grave, hipotonia e defeitos cardíacos. Os recém-nascidos não atingem nenhum marco motor. Muitas vezes, esses bebês apresentam diplegia facial (paralisia facial), falta de reação a estímulos e defeito cardíaco congênito.1,2,3 Os pacientes diagnosticados com AME tipo 0 morrem de insuficiência respiratória aos 6 meses de idade e, por vezes, no primeiro mês após o nascimento.
As crianças que têm sintomas perceptíveis de AME no nascimento ou pouco tempo após o nascimento são geralmente muito fracas; têm dificuldade em respirar, sugar e engolir; e nunca atingem o marco de desenvolvimento da capacidade de se sentarem sozinhas (AME tipo 1 ou doença de Werdnig-Hoffmann). Com tecnologia como a ventilação mecânica e tubos de alimentação para ajudar na respiração e nutrição, as crianças com AME tipo 1 podem sobreviver durante vários anos. Para mais informações sobre o estado atual da pesquisa e novos tratamentos, consulte Pesquisa ou MDA Celebra a Aprovação do FDA de Zolgensma para Tratamento da Atrofia Muscular Espinhal em Pacientes Pediátricos.
SMA tipo 2 (doença de Dubowitz, ou AME intermediária) os sintomas começam em bebês de aproximadamente 3 a 15 meses de idade que aprendem a sentar sem assistência, mas não ficam de pé ou caminham independentemente. Esta apresentação é responsável por cerca de 20% de todos os casos de AME. A fraqueza muscular é predominantemente proximal (perto do centro do corpo) e envolve mais os membros inferiores do que os membros superiores. Normalmente, os músculos da face e dos olhos não são afetados.4 Embora as complicações respiratórias sejam uma ameaça constante, as crianças com AME tipo 2 geralmente vivem até a idade adulta jovem, e muitas vivem mais.5
Quando a fraqueza muscular começa em crianças mais velhas e adolescentes que aprendem a ficar de pé e andar, mas perdem essas habilidades mais tarde na vida, a doença pode ser rotulada como AME tipo 3 (também conhecida como AME leve, início juvenil, ou doença de Kugelberg-Welander). A AME do tipo 3 é responsável por cerca de 30% dos casos de AME. Embora alguns com tipo 3 parem de andar na adolescência, outros caminham bem até à idade adulta. A maioria dos pacientes desenvolve deformidades dos pés, escoliose e fraqueza muscular respiratória.
SMA que surge no final da adolescência ou na idade adulta é chamada de AME tipo 4, ou AME tardia. A esperança de vida é geralmente normal neste tipo. Os pacientes são capazes de atingir todos os marcos motores e manter a sua ambulação ao longo da vida.4
Na SMA tipos 1 a 4, os músculos mais próximos do centro do corpo (músculos proximais) são normalmente mais afectados, ou pelo menos afectados muito mais cedo, do que os músculos mais afastados do centro. Por exemplo, os músculos das coxas são mais fracos do que os músculos das pernas e pés.
 >
>
As pernas tendem a enfraquecer antes dos braços. As mãos podem enfraquecer eventualmente, mas geralmente permanecem mais fortes por mais tempo e, mesmo que enfraqueçam, geralmente permanecem fortes o suficiente para digitar no teclado do computador e outras funções básicas da vida moderna.
O perigo mais sério em AME vem da fraqueza dos músculos necessários para respirar. Uma atenção cuidadosa à função respiratória é necessária ao longo da vida, com pronta atenção às infecções. O seu médico pode ajudá-lo com os detalhes da manutenção da saúde respiratória, incluindo a eliminação de secreções e talvez a ventilação assistida (não necessariamente 24 horas por dia).
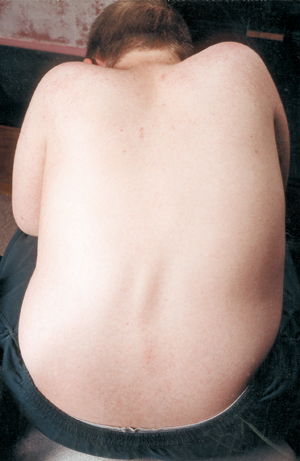
Outra complicação médica na AME é a curvatura da coluna vertebral, normalmente um tipo de curvatura de lado a lado chamada escoliose. A escoliose ocorre devido à fraqueza dos músculos que normalmente suportam a coluna vertebral, que é uma coluna flexível. A escoliose pode ser muito desconfortável, interferir na posição e mobilidade e danificar a imagem corporal de uma criança (ou adulto). Alguns estudos têm mostrado que as curvaturas da coluna vertebral, se forem graves, podem interferir na respiração.
Muitas crianças com AME começam a mostrar uma curva escamosa no início da vida, que muitas vezes é tratada com uma cinta até que seja atingido o momento certo para a cirurgia. Os cirurgiões geralmente gostam de esperar até que o crescimento esteja completo ou quase, antes de endireitar e fundir cirurgicamente a coluna vertebral. Eles também levam em consideração a função pulmonar de uma criança e a probabilidade de progresso da curva espinhal.
SMA não ligado ao cromossomo 5
Algumas formas de SMA não estão ligadas ao cromossomo 5 ou à deficiência de SMN. Estas formas variam muito na severidade e nos músculos mais afetados. Enquanto a maioria das formas, como a forma relacionada ao cromossoma 5, afeta principalmente os músculos proximais, outras formas existem que afetam principalmente os músculos distais, aqueles mais distantes do centro do corpo, pelo menos no início.
- Rudnik-Schöneborn, S. et al. A doença cardíaca congênita é uma característica da atrofia muscular espinhal infantil grave. J. Med. Genet. (2008). doi:10.1136/jmg.2008.057950
- Grotto, S. et al. Tipo 0 Atrofia Muscular Espinhal: Delineação adicional das características pré e pós-natais em 16 pacientes. J. Neuromuscul. Dis. (2016). doi:10.3233/JND-160177
- Menke, L. A. et al. Defeitos cardíacos congênitos na atrofia muscular espinhal tipo I: Um relato clínico de dois irmãos e uma revisão da literatura. Am. J. Med. Genet. Parte A (2008). doi:10.1002/ajmg.a.32233
- Butchbach, M. E. R. Copy Number Variations in the Survival Motor Neuron Genes: Implicações para a Atrofia Muscular Espinhal e Outras Doenças Neurodegenerativas. Frente. Mol. Biosci. (2016). doi:10.3389/fmolb.2016.00007
- Zerres, K. & Schöneborn, S. R. Natural History in Proximal Spinal Muscular Atrophy: Análise Clínica de 445 Pacientes e Sugestões para uma Modificação das Classificações Existentes. Arco. Neurol. (1995). doi:10.1001/archneur.19900540290108025