
Segni e Sintomi
SMA legata al cromosoma 5 (SMN-related), tipi 0-4
Nell’atrofia muscolare spinale (SMA) dai tipi 0 ai 4, i sintomi variano su un continuum da grave a lieve in base alla quantità di proteina SMN funzionale presente nelle cellule nervose chiamate motoneuroni. (“SMN” sta per sopravvivenza del motoneurone) Più proteina SMN c’è, più tardi nella vita iniziano i sintomi e più lieve è il corso della malattia. Il numero di copie dei geni SMN2, che possono compensare il cattivo funzionamento dei geni SMN1, determina la gravità della SMA e l’età di insorgenza del paziente.
Le madri di bambini affetti da SMA tipo 0 possono riferire una diminuzione del movimento fetale alla fine della gravidanza e partorire prima. Questi neonati si presentano con grave debolezza, ipotonia e difetti cardiaci. I neonati non raggiungono alcuna pietra miliare motoria. Spesso questi bambini hanno una diplegia facciale (paralisi del viso), una mancanza di reazione agli stimoli e un difetto cardiaco congenito.1,2,3 I pazienti con diagnosi di SMA tipo 0 muoiono per insufficienza respiratoria entro i 6 mesi di età e talvolta entro il primo mese dalla nascita.
I bambini che hanno sintomi evidenti di SMA alla nascita o poco dopo sono di solito molto deboli; hanno difficoltà a respirare, succhiare e deglutire; e non raggiungono mai la pietra miliare dello sviluppo di essere in grado di sedersi da soli (SMA tipo 1 o malattia di Werdnig-Hoffmann). Con tecnologie come la ventilazione meccanica e tubi di alimentazione per assistere la respirazione e la nutrizione, i bambini con SMA tipo 1 possono sopravvivere per un certo numero di anni. Per ulteriori informazioni sullo stato attuale della ricerca e sui nuovi trattamenti, vedere Ricerca o MDA celebra l’approvazione FDA di Zolgensma per il trattamento dell’atrofia muscolare spinale nei pazienti pediatrici.
I sintomi della SMA di tipo 2 (nota anche come malattia di Dubowitz, o SMA intermedia) iniziano nei bambini a circa 3-15 mesi di età che imparano a sedersi senza assistenza ma non stanno in piedi o camminano autonomamente. Questa presentazione rappresenta circa il 20% di tutti i casi di SMA. La debolezza muscolare è prevalentemente prossimale (vicino al centro del corpo) e coinvolge gli arti inferiori più degli arti superiori. Sebbene le complicazioni respiratorie siano una minaccia costante, i bambini affetti da SMA tipo 2 di solito vivono fino alla giovane età adulta, e molti vivono più a lungo.5
Quando la debolezza muscolare inizia in bambini e ragazzi più grandi che imparano a stare in piedi e a camminare, ma perdono queste abilità più tardi nella vita, la malattia può essere etichettata come SMA tipo 3 (nota anche come SMA lieve, insorgenza giovanile o malattia di Kugelberg-Welander). La SMA tipo 3 rappresenta circa il 30% dei casi di SMA. Anche se alcuni con il tipo 3 smettono di camminare nell’adolescenza, altri camminano bene fino all’età adulta. La maggior parte dei pazienti sviluppa deformità del piede, scoliosi e debolezza dei muscoli respiratori.
La SMA che insorge nella tarda adolescenza o in età adulta è chiamata tipo 4, o SMA a esordio tardivo. La durata della vita è generalmente normale in questo tipo. I pazienti sono in grado di raggiungere tutte le tappe motorie e mantenere la deambulazione per tutta la vita.4
Nei tipi di SMA da 1 a 4, i muscoli più vicini al centro del corpo (muscoli prossimali) di solito sono più colpiti, o almeno colpiti molto prima, dei muscoli più lontani dal centro. Per esempio, i muscoli delle cosce sono più deboli dei muscoli della parte inferiore delle gambe e dei piedi.
Le gambe tendono a indebolirsi prima delle braccia. Le mani possono indebolirsi alla fine, ma di solito rimangono più forti a lungo e, anche se si indeboliscono, di solito rimangono abbastanza forti per digitare sulla tastiera di un computer e altre funzioni di base della vita moderna.
Il pericolo più serio nella SMA deriva dalla debolezza dei muscoli necessari per la respirazione. Un’attenta attenzione alla funzione respiratoria è necessaria per tutta la vita, con una pronta attenzione alle infezioni. Il medico può aiutarvi con i dettagli del mantenimento della salute respiratoria, compresa la rimozione delle secrezioni e forse la ventilazione assistita (non necessariamente 24 ore su 24).
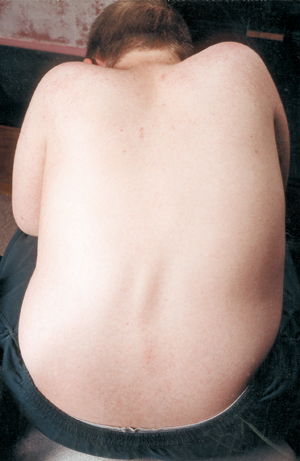
Un’altra complicazione medica nella SMA è la curvatura spinale, di solito un tipo di curvatura laterale chiamato scoliosi. La scoliosi si verifica a causa della debolezza dei muscoli che normalmente sostengono la colonna vertebrale, che è una colonna flessibile. La scoliosi può essere molto scomoda, interferire con la posizione e la mobilità, e danneggiare l’immagine del corpo di un bambino (o di un adulto). Alcuni studi hanno dimostrato che le curvature spinali, se sono gravi, possono interferire con la respirazione.
Molti bambini con SMA iniziano a mostrare una curva scoliotica nei primi anni di vita, che spesso viene trattata con un tutore fino al momento giusto per la chirurgia. I chirurghi generalmente preferiscono aspettare che la crescita sia completa o quasi prima di raddrizzare e fondere chirurgicamente la colonna vertebrale. Prendono anche in considerazione la funzione polmonare del bambino e la velocità con cui la curva spinale è destinata a progredire.
SMA non legata al cromosoma 5
Alcune forme di SMA non sono legate al cromosoma 5 o alla carenza di SMN. Queste forme variano notevolmente nella gravità e nei muscoli più colpiti. Mentre la maggior parte delle forme, come quella legata al cromosoma 5, colpisce soprattutto i muscoli prossimali, esistono altre forme che colpiscono soprattutto i muscoli distali, quelli più lontani dal centro del corpo, almeno all’inizio.
- Rudnik-Schöneborn, S. et al. J. Med. Genet. (2008). doi:10.1136/jmg.2008.057950
- Grotto, S. et al. Atrofia muscolare spinale tipo 0: Ulteriore delineazione delle caratteristiche prenatali e postnatali in 16 pazienti. J. Neuromuscul. Dis. (2016). doi:10.3233/JND-160177
- Menke, L. A. et al. Congenital heart defects in spinal muscular atrophy type I: Un rapporto clinico di due fratelli e una revisione della letteratura. Am. J. Med. Genet. Part A (2008). doi:10.1002/ajmg.a.32233
- Butchbach, M. E. R. Copy Number Variations in the Survival Motor Neuron Genes: Implicazioni per l’atrofia muscolare spinale e altre malattie neurodegenerative. Fronte. Mol. Biosci. (2016). doi:10.3389/fmolb.2016.00007
- Zerres, K. & Schöneborn, S. R. Natural History in Proximal Spinal Muscular Atrophy: Analisi clinica di 445 pazienti e suggerimenti per una modifica delle classificazioni esistenti. Arch. Neurol. (1995). doi:10.1001/archneur.19900540290108025