
Tuntomerkit ja oireet
SMA liittyy kromosomiin 5 (SMN:ään liittyvä), tyypit 0-4
Tyypeissä 0-4 selkäydinlihasatrofiassa (SMA) oireet vaihtelevat vaikeasta lievään riippuen siitä, kuinka paljon toimivaa SMN-proteiinia on hermosoluissa, joita kutsutaan liikkeellepanoneuroneiksi. (”SMN” tulee sanoista survival of motor neuron.) Mitä enemmän SMN-proteiinia on, sitä myöhemmin elämässä oireet alkavat ja sitä lievempi taudin kulku on. SMN2-geenien, jotka voivat kompensoida SMN1-geenien toimintahäiriöitä, kopioiden lukumäärä määrittää SMA:n vaikeusasteen ja potilaan puhkeamisiän.
Tyypin 0 SMA:ta sairastavien lasten äidit saattavat raportoida sikiön liikkeiden vähenemisestä raskauden loppuvaiheessa ja synnyttää aikaisemmin. Näillä vastasyntyneillä on vaikeaa heikkoutta, hypotoniaa ja sydänvikoja. Vastasyntyneet eivät saavuta mitään motorisia virstanpylväitä. Usein näillä vauvoilla on kasvojen diplegia (kasvohalvaus), reagoimattomuus ärsykkeisiin ja synnynnäinen sydänvika1,2,3. Tyypin 0 SMA:ta sairastavat potilaat kuolevat hengitysvajaukseen kuuden kuukauden ikään mennessä ja joskus jo ensimmäisen kuukauden kuluessa syntymästä.
Lapset, joilla on havaittavissa SMA:n oireita syntymähetkellä tai pian syntymän jälkeen, ovat yleensä hyvin heikkoja, heillä on hengitys-, imemis- ja nielemisvaikeuksia eivätkä he koskaan saavuta kehityksen virstanpylvästä, jolloin he pystyvät istumaan itsenäisesti (SMA-tyypin 1 tai Werdnig-Hoffmannin tauti). Teknologian, kuten mekaanisen hengityskoneiston ja syöttöletkujen avulla hengityksen ja ravitsemuksen tukemiseksi SMA-tyypin 1 lapset voivat selvitä hengissä useita vuosia. Lisätietoja tutkimuksen nykytilasta ja uusista hoidoista on kohdassa Tutkimus tai MDA Celebrates FDA Approval of Zolgensma for the Treatment of Spinal Muscular Atrophy in Pediatric Patients (MDA juhlii Zolgensman FDA-hyväksyntää selkäydinlihasatrofian hoitoon pediatrisilla potilailla). Tämä taudinkuva on noin 20 prosenttia kaikista SMA-tapauksista. Lihasheikkous on pääasiassa proksimaalista (lähellä kehon keskiosaa) ja koskee alaraajoja enemmän kuin yläraajoja. Yleensä kasvojen ja silmien lihakset eivät ole vaurioituneet.4 Vaikka hengityskomplikaatiot ovat jatkuva uhka, tyypin 2 SMA:ta sairastavat lapset elävät yleensä nuoreen aikuisikään asti, ja monet elävät pidempäänkin.5
Kun lihasheikkous alkaa vanhemmilla lapsilla ja teini-ikäisillä, jotka oppivat seisomaan ja kävelemään, mutta menettävät nämä kyvyt myöhemmin elämässään, tautia saatetaan nimittää tyypin 3 SMA:ksi (eli lieväksi SMA:ksi, nuoruusiän alkavaksi SMA-oireyhtymäksi (juvenile onset disease) tai Kugelbergin-Welanderin taudiksi). SMA-tyypin 3 osuus SMA-tapauksista on noin 30 prosenttia. Vaikka jotkut tyypin 3 potilaat lakkaavat kävelemästä teini-iässä, toiset kävelevät pitkälle aikuisikään asti. Useimmille potilaille kehittyy jalkojen epämuodostumia, skolioosia ja hengityslihasten heikkoutta.
SMA:ta, joka puhkeaa myöhäisessä teini-iässä tai aikuisuudessa, kutsutaan tyypiksi 4 eli myöhäis-SMA:ksi. Elinikä on tässä tyypissä yleensä normaali. Potilaat pystyvät saavuttamaan kaikki motoriset virstanpylväät ja säilyttävät kävelykykynsä koko elämänsä ajan.4
SMA-tyypeissä 1-4 lähempänä kehon keskipistettä olevat lihakset (proksimaaliset lihakset) sairastuvat tavallisesti enemmän tai ainakin sairastuvat paljon aikaisemmin kuin kauempana keskipisteestä olevat lihakset. Esimerkiksi reisien lihakset ovat heikompia kuin alaraajojen ja jalkaterien lihakset.
Jalkojen lihakset heikkenevät yleensä ennen käsiä. Kädet voivat lopulta heikentyä, mutta ne pysyvät yleensä vahvimpina pisimpään, ja vaikka ne heikkenisivätkin, ne pysyvät yleensä riittävän vahvoina tietokoneen näppäimistöllä kirjoittamiseen ja muihin nykyelämän perustoimintoihin.
Suurimman vaaran SMA:ssa aiheuttaa hengitykseen tarvittavien lihasten heikkous. Hengitystoimintaan on kiinnitettävä huolellista huomiota koko elämän ajan, ja infektioihin on kiinnitettävä nopeasti huomiota. Lääkäri voi auttaa sinua hengityselinten terveyden ylläpitämiseen liittyvissä yksityiskohdissa, mukaan lukien eritteiden puhdistaminen ja ehkä avustettu ventilaatio (ei välttämättä ympärivuorokautisesti).
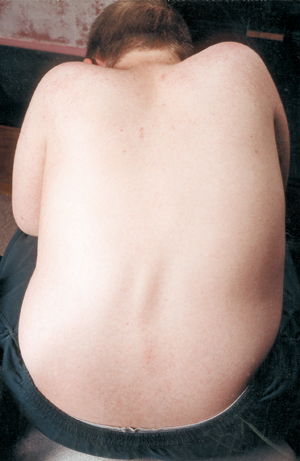
Muutama lääketieteellinen komplikaatio SMA:ssa on selkärangan kaarevuus, yleensä sivulta toiselle suuntautuva kaarevuustyyppi, jota kutsutaan skolioosiksi. Skolioosi johtuu niiden lihasten heikkoudesta, jotka normaalisti tukevat selkärankaa, joka on joustava pylväs. Skolioosi voi olla hyvin epämiellyttävä, haitata asentoa ja liikkuvuutta ja vahingoittaa lapsen (tai aikuisen) kehonkuvaa. Jotkut tutkimukset ovat osoittaneet, että selkärangan kaarevuus, jos se on vakava, voi häiritä hengitystä.
Monilla SMA:ta sairastavilla lapsilla skolioottinen käyrä alkaa näkyä jo varhain elämässä, ja sitä hoidetaan usein tukivarsilla, kunnes oikea aika leikkaukseen on saavutettu. Kirurgit haluavat yleensä odottaa, kunnes kasvu on päättynyt tai lähes päättynyt, ennen kuin selkäranka oikaistaan ja fuusioidaan kirurgisesti. He ottavat huomioon myös lapsen keuhkojen toiminnan ja sen, kuinka nopeasti selkärangan käyrä todennäköisesti etenee.
SMA ei liity kromosomiin 5
Jotkut SMA:n muodot eivät liity kromosomiin 5 tai SMN:n puutokseen. Nämä muodot vaihtelevat suuresti vaikeusasteeltaan ja eniten kärsivien lihasten suhteen. Vaikka useimmat muodot, kuten kromosomiin 5 liittyvä muoto, vaikuttavat enimmäkseen proksimaalisiin lihaksiin, on olemassa muitakin muotoja, jotka vaikuttavat enimmäkseen distaalisiin lihaksiin, eli niihin, jotka ovat ainakin alussa kauempana kehon keskipisteestä.
- Rudnik-Schöneborn, S. et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J. Med. Genet. (2008). doi:10.1136/jmg.2008.057950
- Grotto, S. et al. Type 0 Spinal Muscular Atrophy: Further Delineation of Prenatal and Postnatal Features in 16 Patients. J. Neuromuscul. Dis. (2016). doi:10.3233/JND-160177
- Menke, L. A. et al. Congenital heart defects in spinal muscular atrophy type I: Kliininen raportti kahdesta sisaruksesta ja kirjallisuuskatsaus. Am. J. Med. Genet. Part A (2008). doi:10.1002/ajmg.a.32233
- Butchbach, M. E. R. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front. Mol. Biosci. (2016). doi:10.3389/fmolb.2016.00007
- Zerres, K. & Schöneborn, S. R. Natural History in Proximal Spinal Muscular Atrophy: Clinical Analysis of 445 Patients and Suggestions for a Modification of Existing Classifications. Arch. Neurol. (1995). doi:10.1001/archneur.19900540290108025
.