
Objawy i symptomy
SMA związane z chromosomem 5 (SMN-related), typy 0-4
W rdzeniowym zaniku mięśni (SMA) typu 0 do 4, objawy zmieniają się na kontinuum od ciężkich do łagodnych w zależności od ilości funkcjonalnego białka SMN w komórkach nerwowych zwanych neuronami ruchowymi. („SMN” oznacza przetrwanie neuronu ruchowego.) Im więcej białka SMN, tym później w życiu zaczynają się objawy i tym łagodniejszy jest przebieg choroby. Liczba kopii genów SMN2, które mogą kompensować nieprawidłowo działające geny SMN1, decyduje o ciężkości SMA i wieku wystąpienia choroby.
Matki dzieci dotkniętych SMA typu 0 mogą zgłaszać zmniejszenie ruchów płodu w późnym okresie ciąży i rodzić wcześniej. Noworodki te prezentują się z ciężkim osłabieniem, hipotonią i wadami serca. Noworodki nie osiągają żadnych kamieni milowych w rozwoju ruchowym. Często u tych dzieci występuje diplegia twarzy (paraliż twarzy), brak reakcji na bodźce oraz wrodzona wada serca.1,2,3 Pacjenci, u których zdiagnozowano SMA typu 0, umierają z powodu niewydolności oddechowej w wieku 6 miesięcy, a czasami w ciągu pierwszego miesiąca po urodzeniu.
Dzieci, u których zauważono objawy SMA w chwili urodzenia lub wkrótce po urodzeniu, są zazwyczaj bardzo słabe; mają trudności z oddychaniem, ssaniem i połykaniem; i nigdy nie osiągają kamienia milowego w rozwoju, jakim jest zdolność do samodzielnego siedzenia (SMA typu 1 lub choroba Werdniga-Hoffmanna). Dzięki takim technologiom, jak wentylacja mechaniczna i rurki do karmienia wspomagające oddychanie i odżywianie, dzieci z SMA typu 1 mogą przeżyć wiele lat. Więcej informacji na temat aktualnego stanu badań i nowych metod leczenia można znaleźć w części Badania naukowe lub MDA Celebrates FDA Approval of Zolgensma for Treatment of Spinal Muscular Atrophy in Pediatric Patients.
Objawy SMA typu 2 (inaczej choroby Dubowitza lub pośredniego SMA) rozpoczynają się u niemowląt w wieku od około 3 do 15 miesięcy, które uczą się siedzieć bez pomocy, ale nie stoją ani nie chodzą samodzielnie. Ta postać choroby stanowi około 20% wszystkich przypadków SMA. Osłabienie mięśni jest przeważnie proksymalne (blisko środka ciała) i dotyczy bardziej kończyn dolnych niż górnych. Zazwyczaj mięśnie twarzy i oczu są nienaruszone.4 Chociaż komplikacje oddechowe są stałym zagrożeniem, dzieci z SMA typu 2 zazwyczaj dożywają późnej dorosłości, a wiele z nich żyje dłużej.5
Gdy osłabienie mięśni zaczyna się u starszych dzieci i nastolatków, którzy uczą się stać i chodzić, ale tracą te umiejętności w późniejszym okresie życia, choroba może być określana jako SMA typu 3 (określane jako łagodne SMA, początek młodzieńczy lub choroba Kugelberga-Welandera). SMA typu 3 stanowi około 30% przypadków SMA. Chociaż niektórzy pacjenci z typem 3 przestają chodzić w okresie dojrzewania, inni chodzą nawet w wieku dojrzałym. U większości pacjentów występują zniekształcenia stóp, skolioza i osłabienie mięśni oddechowych.
SMA, które pojawia się w późnych latach młodzieńczych lub w wieku dorosłym, nazywane jest typem 4 lub SMA o późnym początku. W tym typie choroby długość życia jest na ogół normalna. Pacjenci są w stanie osiągnąć wszystkie kamienie milowe rozwoju motorycznego i zachować zdolność poruszania się przez całe życie.4
W SMA typu 1 do 4 mięśnie znajdujące się bliżej środka ciała (mięśnie proksymalne) są zwykle bardziej dotknięte chorobą, lub przynajmniej są nią dotknięte znacznie wcześniej, niż mięśnie znajdujące się dalej od środka. Na przykład, mięśnie ud są słabsze niż mięśnie dolnej części nóg i stóp.
Nogi mają tendencję do osłabiania się przed ramionami. Ręce mogą w końcu osłabnąć, ale zwykle pozostają najsilniejsze najdłużej, a nawet jeśli osłabną, zwykle pozostają wystarczająco silne do pisania na klawiaturze komputera i innych podstawowych funkcji współczesnego życia.
Najpoważniejsze zagrożenie w SMA wynika z osłabienia mięśni niezbędnych do oddychania. Przez całe życie należy zwracać baczną uwagę na funkcjonowanie układu oddechowego i szybko reagować na infekcje. Lekarz może pomóc w ustaleniu szczegółów dotyczących utrzymania zdrowia układu oddechowego, w tym usuwania wydzieliny i być może wspomaganej wentylacji (niekoniecznie całodobowej).
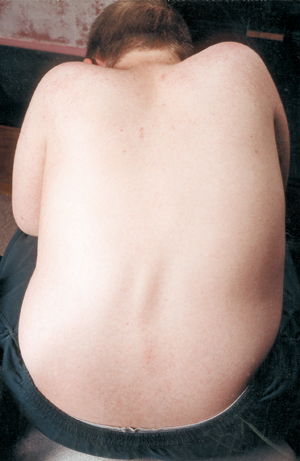
Innym powikłaniem medycznym w SMA jest skrzywienie kręgosłupa, zwykle w kierunku bocznym, zwane skoliozą. Skolioza występuje z powodu osłabienia mięśni, które normalnie podtrzymują kręgosłup, będący elastyczną kolumną. Skolioza może być bardzo niewygodna, zaburzać pozycję i mobilność, a także szkodzić obrazowi ciała dziecka (lub osoby dorosłej). Niektóre badania wykazały, że skrzywienia kręgosłupa, jeśli są poważne, mogą przeszkadzać w oddychaniu.
Wiele dzieci z SMA zaczyna wykazywać skrzywienie skoliotyczne we wczesnym okresie życia, co często jest leczone za pomocą ortez, aż do osiągnięcia odpowiedniego czasu na operację. Chirurdzy na ogół lubią czekać z operacyjnym wyprostowaniem i zespoleniem kręgosłupa do czasu zakończenia wzrostu lub prawie zakończenia. Biorą również pod uwagę wydolność płuc dziecka i to, jak szybko krzywizna kręgosłupa może się rozwijać.
SMA niezwiązane z chromosomem 5
Niektóre postacie SMA nie są związane z chromosomem 5 ani z niedoborem SMN. Formy te różnią się znacznie pod względem ciężkości i najbardziej dotkniętych mięśni. Podczas gdy większość form, jak forma związana z chromosomem 5, dotyka głównie mięśni proksymalnych, istnieją inne formy, które dotykają głównie mięśni dystalnych, tych bardziej oddalonych od centrum ciała, przynajmniej na początku.
- Rudnik-Schöneborn, S. et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J. Med. Genet. (2008). doi:10.1136/jmg.2008.057950
- Grotto, S. et al. Type 0 Spinal Muscular Atrophy: Further Delineation of Prenatal and Postnatal Features in 16 Patients. J. Neuromuscul. Dis. (2016). doi:10.3233/JND-160177
- Menke, L. A. et al. Congenital heart defects in spinal muscular atrophy type I: A clinical report of two siblings and a review of the literature. Am. J. Med. Genet. Part A (2008). doi:10.1002/ajmg.a.32233
- Butchbach, M. E. R. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front. Mol. Biosci. (2016). doi:10.3389/fmolb.2016.00007
- Zerres, K. & Schöneborn, S. R. Natural History in Proximal Spinal Muscular Atrophy: Clinical Analysis of 445 Patients and Suggestions for a Modification of Existing Classifications. Arch. Neurol. (1995). doi:10.1001/archneur.19900540290108025
.