
Lizosomy są kulistymi, związanymi z błoną organellami, które są generowane przez aparat Golgiego. Zawierają one enzymy hydrolityczne, dzięki czemu funkcjonują jako część systemu recyklingu komórki.
W tym artykule przyjrzymy się strukturze, syntezie i funkcji lizosomów oraz rozważymy ich znaczenie dla praktyki klinicznej.
Struktura
Lizosomy są kwaśnymi organellami związanymi z błoną, występującymi w komórkach, zwykle o długości około 1 mikrometra. Lizosomy zawierają liczne enzymy hydrolityczne, które katalizują reakcje hydrolizy.
Błona otaczająca lizosom jest niezbędna do zapewnienia, że enzymy te nie wyciekną do cytoplazmy i nie uszkodzą komórki od wewnątrz. W celu utrzymania kwaśnego pH lizosomu, protony są aktywnie transportowane do organelli przez błonę lizosomalną.
Synteza
Lizosom i enzymy w nim zawarte są syntetyzowane oddzielnie. Białka lizosomalne powstają w taki sam sposób jak każde inne białko. Pierwszym etapem jest zainicjowanie produkcji nici mRNA z odpowiednich odcinków DNA. Nici mRNA przechodzą do szorstkiego retikulum endoplazmatycznego, gdzie rybosomy konstruują enzymy hydrolityczne.
Co ważne, są one znakowane mannozo-6-fosforanem w aparacie Golgiego, aby skierować je do lizosomu. W rezultacie pęcherzyki zawierające te enzymy odrywają się od aparatu Golgiego. Za przyłączenie znacznika mannozy-6-fosforanu odpowiedzialne są dwa enzymy: fosfotransferaza N-acetyloglukozaminy i fosfoglikozydaza N-acetyloglukozaminy.
Weseczka ta, znajdująca się teraz w cytoplazmie, łączy się następnie z późnym endosomem, który jest kolejną kwaśną, błoniastą organellą. Późny endosom ma pompy protonowe w swojej błonie, które utrzymują jego wewnętrzne środowisko kwaśne. Niskie pH powoduje dysocjację białka od receptora mannozy-6-fosforanu. Receptor ten może być następnie zawrócony z powrotem do aparatu Golgiego.
Grupa fosforanowa jest również usuwana ze znacznika mannozy-6-fosforanu, aby zapobiec powrotowi całego białka do aparatu Golgiego. Późny endosom może ostatecznie dojrzeć do lizosomu, po otrzymaniu enzymów z aparatu Golgiego.
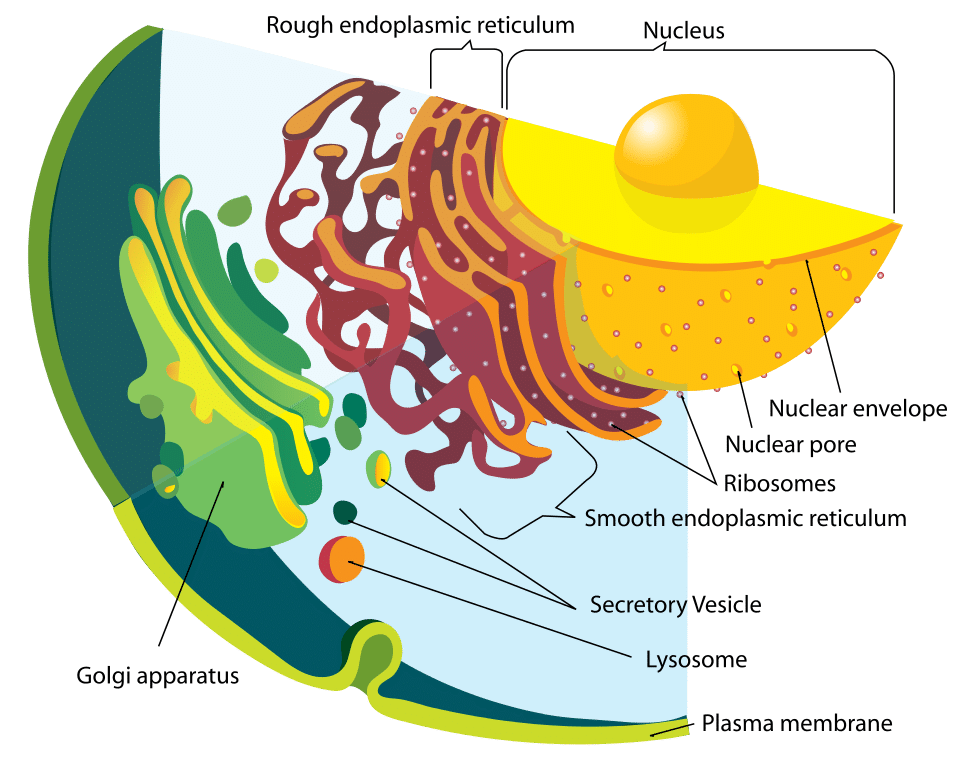 Rysunek 1 – Schemat systemu endomembranowego
Rysunek 1 – Schemat systemu endomembranowegoFunkcja
Enzymy hydrolityczne zawarte w lizosomie pozwalają na zniszczenie obcych cząstek. Lizosomy odgrywają ważną rolę w fagocytozie. Kiedy makrofagi fagocytują obce cząstki, zawierają je w fagosomie. Następnie fagosom łączy się z lizosomem, tworząc fagolizosom.
Ezymy te są krytyczne w mechanizmach zabijania niezależnych od tlenu. Lizosomy pomagają również bronić się przed wnikaniem patogenów poprzez endocytozę, degradując patogeny zanim dotrą do cytoplazmy.
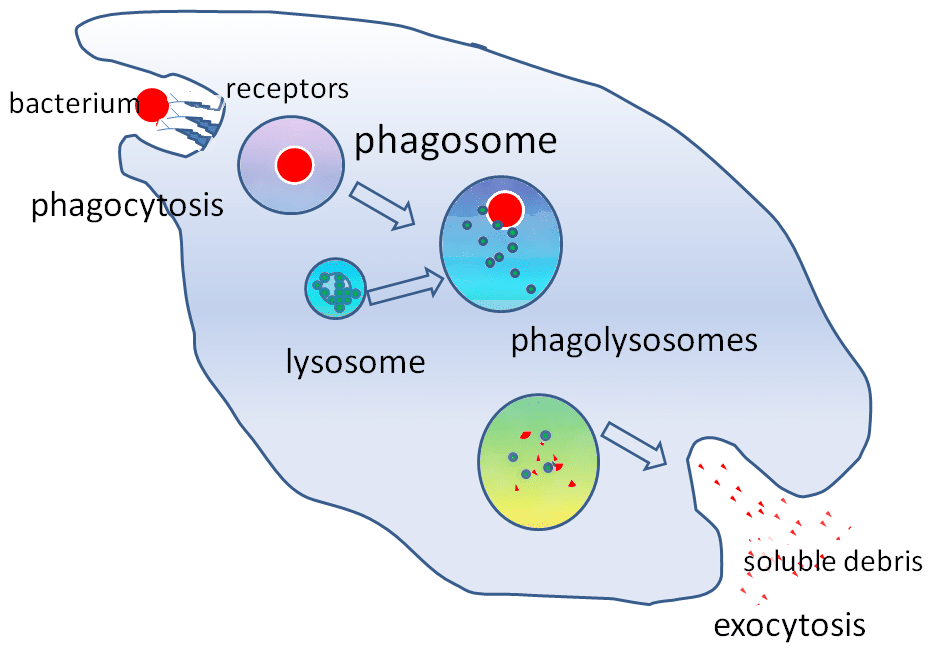 Rycina 2 – Rola lizosomu w fagocytozie
Rycina 2 – Rola lizosomu w fagocytozieWażność kliniczna
Choroba komórek I
Jest ona spowodowana defektami genetycznymi w enzymie fosfotransferazy N-acetyloglukozaminy. Enzym ten jest niezbędny do dodawania mannozy-6-fosforanu do białek kierowanych do lizosomu. Powoduje to, że enzymy lizosomalne nie są właściwie ukierunkowane. W rezultacie znaczne ich ilości znajdują się zarówno w moczu, jak i w krwiobiegu.
Lizosomalne choroby spichrzeniowe
Jest to grupa schorzeń genetycznych wpływających na lizosomy. Choroby te różnią się znacznie pod względem oznak, objawów i danych demograficznych pacjentów. Istnieje kilka klasyfikacji; najczęstszym z tych schorzeń jest choroba Gauchera. Jest ona spowodowana niedoborem beta-glukocerebrozydazy. Enzym ten jest wymagany do rozkładu glukocerebrozydu. Bez tego enzymu, glukocerebrozyd jest w stanie gromadzić się w komórkach, co może powodować ich uszkodzenie. Objawy obejmują hepatosplenomegalię i anemię.
.