
Tekenen en Symptomen
SMA gekoppeld aan chromosoom 5 (SMN-gerelateerd), typen 0-4
Bij spinale musculaire atrofie (SMA), typen 0 tot en met 4, variëren de symptomen op een continuüm van ernstig tot mild, afhankelijk van hoeveel functioneel SMN-eiwit er aanwezig is in de zenuwcellen die motorneuronen worden genoemd. (“SMN” staat voor survival of motor neuron.) Hoe meer SMN-eiwit er is, hoe later in het leven de symptomen beginnen en hoe milder het verloop van de ziekte. Het aantal kopieën van SMN2 genen, die slecht functionerende SMN1 genen kunnen compenseren, bepaalt de ernst van SMA en de leeftijd waarop de patiënt de ziekte krijgt.
Moeders van kinderen met SMA type 0 kunnen aan het eind van de zwangerschap een afname in foetale beweging melden en eerder bevallen. Deze pasgeborenen presenteren zich met ernstige zwakte, hypotonie, en hartafwijkingen. Pasgeborenen bereiken geen motorische mijlpalen. Vaak hebben deze baby’s diplegie (gezichtsverlamming), een gebrek aan reactie op prikkels, en een aangeboren hartafwijking.1,2,3 Patiënten met SMA type 0 sterven aan ademhalingsmoeilijkheden als ze 6 maanden oud zijn en soms al binnen de eerste maand na de geboorte.
Kinderen die bij of kort na de geboorte duidelijke SMA-symptomen vertonen, zijn meestal erg zwak; hebben moeite met ademen, zuigen en slikken; en bereiken nooit de ontwikkelingsmijlpaal van zelfstandig kunnen zitten (SMA type 1 of de ziekte van Werdnig-Hoffmann). Met behulp van technologie zoals mechanische beademing en voedingsslangen om te helpen bij de ademhaling en de voeding, kunnen kinderen met SMA type 1 nog een aantal jaren overleven. Voor meer informatie over de huidige stand van het onderzoek en nieuwe behandelingen, zie Onderzoek of MDA viert FDA-goedkeuring van Zolgensma voor de behandeling van spinale spieratrofie bij pediatrische patiënten.
SMA type 2 (ook bekend als de ziekte van Dubowitz, of intermediaire SMA) symptomen beginnen bij baby’s van ongeveer 3 tot 15 maanden oud die leren zonder hulp te zitten, maar niet zelfstandig kunnen staan of lopen. Deze presentatie vertegenwoordigt ongeveer 20% van alle gevallen van SMA. De spierzwakte is overwegend proximaal (dicht bij het centrum van het lichaam) en betreft de onderste ledematen meer dan de bovenste ledematen. Meestal zijn het gezicht en de oogspieren niet aangetast.4 Hoewel ademhalingscomplicaties een constante bedreiging zijn, leven kinderen met SMA type 2 meestal tot jongvolwassenheid, en velen leven langer.5
Wanneer de spierzwakte begint bij oudere kinderen en tieners die leren staan en lopen, maar deze vaardigheden later in het leven verliezen, kan de ziekte worden aangeduid als SMA type 3 (ook bekend als milde SMA, juveniele aanvang, of de ziekte van Kugelberg-Welander). SMA type 3 komt voor in ongeveer 30% van de gevallen van SMA. Hoewel sommigen met type 3 stoppen met lopen in de adolescentie, lopen anderen tot ver in hun volwassen jaren. De meeste patiënten ontwikkelen voetmisvormingen, scoliose en ademhalingsspierzwakte.
SMA die in de late tienerjaren of op volwassen leeftijd ontstaat, wordt type 4 genoemd, of late SMA. De levensduur is over het algemeen normaal bij dit type. Patiënten kunnen alle motorische mijlpalen bereiken en hun hele leven blijven lopen.4
Bij SMA type 1 tot en met 4 zijn de spieren die zich dichter bij het centrum van het lichaam bevinden (proximale spieren) meestal meer, of in ieder geval veel eerder, aangedaan dan de spieren die zich verder van het centrum bevinden. De spieren van de dijen zijn bijvoorbeeld zwakker dan de spieren van de onderbenen en voeten.
De benen verzwakken meestal eerder dan de armen. De handen kunnen uiteindelijk verzwakken, maar zij blijven meestal het langst sterk en zelfs als zij verzwakken, blijven zij meestal sterk genoeg voor het typen op een computertoetsenbord en andere basisfuncties van het moderne leven.
Het ernstigste gevaar bij SMA komt door de zwakte van de spieren die nodig zijn voor de ademhaling. Zorgvuldige aandacht voor de ademhalingsfunctie is gedurende het hele leven nodig, met onmiddellijke aandacht voor infecties. Uw arts kan u helpen met de details van het gezond houden van de luchtwegen, inclusief het opruimen van afscheidingen en misschien geassisteerde beademing (niet noodzakelijkerwijs 24 uur per dag).
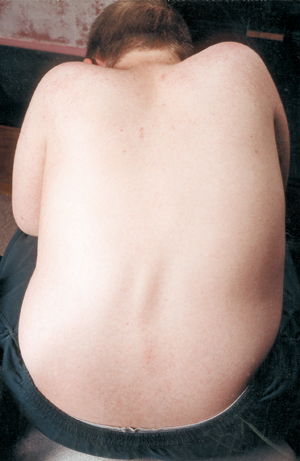
Een andere medische complicatie bij SMA is kromming van de ruggengraat, meestal een kromming van links naar rechts die scoliose wordt genoemd. Scoliose ontstaat door zwakte van de spieren die normaal gesproken de wervelkolom ondersteunen, die een flexibele kolom is. Scoliose kan erg ongemakkelijk zijn, de houding en mobiliteit belemmeren en het lichaamsbeeld van een kind (of volwassene) schaden. Sommige studies hebben aangetoond dat een ernstige verkromming van de wervelkolom de ademhaling kan belemmeren.
Veel kinderen met SMA beginnen al vroeg in hun leven een scoliotische kromming te vertonen, die vaak met een brace wordt behandeld totdat het juiste moment voor een operatie is bereikt. Chirurgen wachten over het algemeen met het operatief rechtzetten en samensmelten van de wervelkolom tot het kind volledig of bijna volledig is gegroeid. Ze houden ook rekening met de longfunctie van een kind en hoe snel de wervelkolomkromming zich waarschijnlijk zal ontwikkelen.
SMA niet gekoppeld aan chromosoom 5
Sommige vormen van SMA zijn niet gekoppeld aan chromosoom 5 of SMN-deficiëntie. Deze vormen variëren sterk in ernst en in de meest aangetaste spieren. Terwijl de meeste vormen, zoals de chromosoom 5-gerelateerde vorm, vooral de proximale spieren aantasten, bestaan er andere vormen die vooral de distale spieren aantasten, die verder weg liggen van het centrum van het lichaam, althans in het begin.
- Rudnik-Schöneborn, S. et al. Congenitale hartziekte is een kenmerk van ernstige infantiele spinale musculaire atrofie. J. Med. Genet. (2008). doi:10.1136/jmg.2008.057950
- Grotto, S. et al. Type 0 Spinal Muscular Atrophy: Further Delineation of Prenatal and Postnatal Features in 16 Patients. J. Neuromuscul. Dis. (2016). doi:10.3233/JND-160177
- Menke, L. A. et al. Congenitale hartafwijkingen bij spinale musculaire atrofie type I: Een klinisch verslag van twee broers en zussen en een overzicht van de literatuur. Am. J. Med. Genet. Part A (2008). doi:10.1002/ajmg.a.32233
- Butchbach, M. E. R. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front. Mol. Biosci. (2016). doi:10.3389/fmolb.2016.00007
- Zerres, K. & Schöneborn, S. R. Natural History in Proximal Spinal Muscular Atrophy: Clinical Analysis of 445 Patients and Suggestions for a Modification of Existing Classifications. Arch. Neurol. (1995). doi:10.1001/archneur.19900540290108025