
Lysosomen zijn bolvormige, membraangebonden organellen die door het golgi-apparaat worden aangemaakt. Ze bevatten hydrolytische enzymen, en fungeren zo als onderdeel van het recyclingsysteem van de cel.
In dit artikel bekijken we de structuur, de synthese en de functie van lysosomen, en bekijken we hun relevantie voor de klinische praktijk.
Structuur
Lysosomen zijn zure, membraangebonden organellen die in cellen worden aangetroffen, gewoonlijk ongeveer 1 micrometer in lengte. Lysosomen bevatten talrijke hydrolytische enzymen die hydrolysereacties katalyseren.
Het membraan dat het lysosoom omgeeft is van vitaal belang om ervoor te zorgen dat deze enzymen niet naar buiten lekken naar het cytoplasma en de cel van binnenuit beschadigen. Om de zure pH van het lysosoom te handhaven, worden protonen actief via het lysosomale membraan in het organel getransporteerd.
Synthese
Het lysosoom en de enzymen erin worden afzonderlijk gesynthetiseerd. Lysosomale proteïnen worden op dezelfde wijze gevormd als alle andere proteïnen. De eerste stap is het op gang brengen van de productie van mRNA-strengen uit relevante DNA-segmenten. De mRNA-strengen gaan naar het ruwe endoplasmatische reticulum, waar ribosomen de hydrolytische enzymen bouwen.
Het is belangrijk dat deze in het Golgi-apparaat met mannose-6-fosfaat worden gelabeld om ze naar het lysosoom te leiden. Het resultaat is dat blaasjes die deze enzymen bevatten, loskomen van het Golgi-apparaat. Twee enzymen zijn verantwoordelijk voor de hechting van het mannose-6-fosfaat-tag: N-acetylglucosamine fosfotransferase en N-acetylglucosamine fosfoglycosidase.
Dit blaasje, nu in het cytoplasma, bindt zich vervolgens aan een laat endosoom, dat een ander zuur, membraangebonden organel is. Het late endosoom heeft protonpompen binnen zijn membraan die zijn interne milieu zuur houden. De lage pH veroorzaakt dissociatie van het eiwit met de mannose-6-fosfaatreceptor. Deze receptor kan dan worden gerecycled terug naar het Golgi-apparaat.
De fosfaatgroep wordt ook verwijderd van de mannose-6-fosfaat tag, om te voorkomen dat het hele eiwit terugkeert naar het Golgi-apparaat. Het late endosoom kan uiteindelijk uitgroeien tot een lysosoom, nadat het de enzymen van het Golgi-apparaat heeft ontvangen.
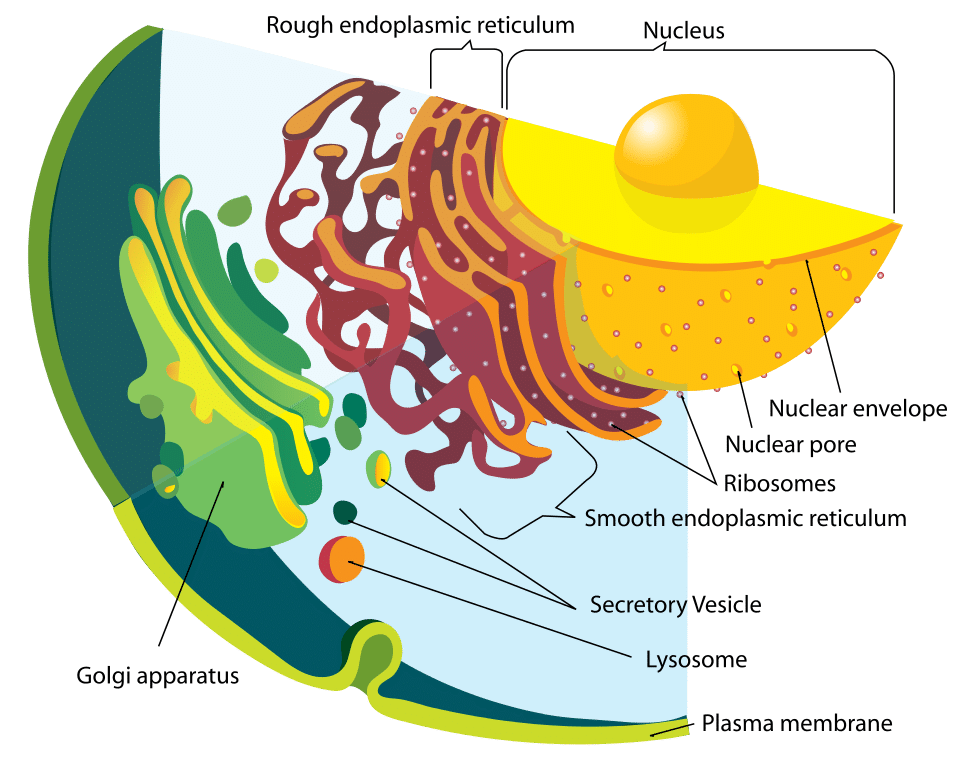 Figuur 1 – Schema van het endomembraanstelsel
Figuur 1 – Schema van het endomembraanstelselFunctie
De hydrolytische enzymen die zich in het lysosoom bevinden, maken het mogelijk vreemde deeltjes te vernietigen. Lysosomen spelen een belangrijke rol bij fagocytose. Wanneer macrofagen vreemde deeltjes fagocytoseren, houden zij deze vast in een fagosoom. Het fagosoom bindt zich dan met een lysosoom om een fagolysosoom te vormen.
Deze enzymen zijn van cruciaal belang bij zuurstofonafhankelijke dodingsmechanismen. Lysosomen helpen ook bij de verdediging tegen het binnendringen van pathogenen via endocytose door pathogenen af te breken voordat zij het cytoplasma bereiken.
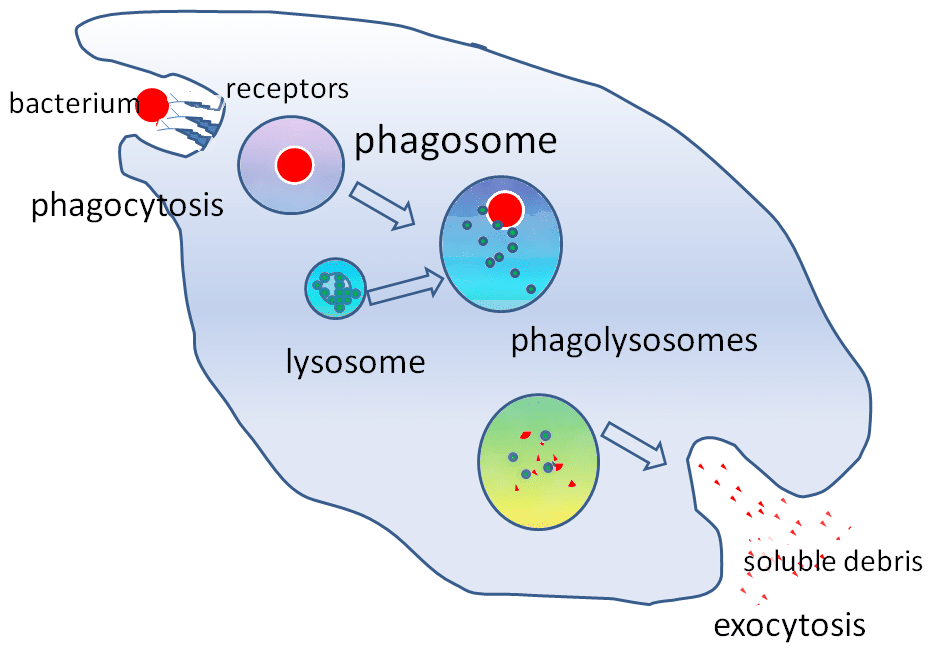 Figuur 2 – De rol van het lysosoom bij fagocytose
Figuur 2 – De rol van het lysosoom bij fagocytoseClinische relevantie
I-celziekte
Dit wordt veroorzaakt door genetische defecten in het N-acetylglucosamine fosfotransferase enzym. Dit enzym is van vitaal belang voor de toevoeging van mannose-6-fosfaat aan lysosoom-gemerkte eiwitten. Dit leidt ertoe dat lysosomale enzymen niet goed gericht zijn. Als gevolg hiervan worden aanzienlijke hoeveelheden gevonden in zowel de urine als de bloedstroom.
Lysosomal Storage Disease
Dit is een groep van genetische aandoeningen die de lysosomen aantasten. De aandoening varieert sterk in tekenen, symptomen en demografie van de patiënt. Er zijn verschillende classificaties; de meest voorkomende van deze aandoeningen is de ziekte van Gaucher. Deze wordt veroorzaakt door een tekort aan bèta-glucocerebrosidase. Dit enzym is nodig om glucocerebroside af te breken. Zonder dit enzym kan het glucocerebroside zich ophopen in de cellen, waardoor deze beschadigd kunnen raken. Symptomen zijn onder meer hepatosplenomegalie en bloedarmoede.