
Los lisosomas son orgánulos esféricos, unidos a la membrana, que son generados por el aparato de golgi. Contienen enzimas hidrolíticas, por lo que funcionan como parte del sistema de reciclaje de la célula.
En este artículo, veremos la estructura, la síntesis y la función de los lisosomas, y consideraremos su relevancia para la práctica clínica.
Estructura
Los lisosomas son orgánulos ácidos unidos a la membrana que se encuentran en el interior de las células y que suelen tener una longitud de alrededor de 1 micrómetro. Los lisosomas contienen numerosas enzimas hidrolíticas que catalizan las reacciones de hidrólisis.
La membrana que rodea al lisosoma es vital para garantizar que estas enzimas no se filtren al citoplasma y dañen la célula desde dentro. Para mantener el pH ácido del lisosoma, los protones se transportan activamente al interior del orgánulo a través de la membrana lisosomal.
Síntesis
El lisosoma y las enzimas que contiene se sintetizan por separado. Las proteínas lisosomales se forman de la misma manera que cualquier otra proteína. El primer paso es la iniciación de la producción de la cadena de ARNm a partir de los segmentos de ADN correspondientes. Las cadenas de ARNm pasan al retículo endoplásmico rugoso, donde los ribosomas construyen las enzimas hidrolíticas.
Es importante destacar que éstas se marcan con manosa-6-fosfato dentro del aparato de Golgi para dirigirlas al lisosoma. Como resultado, las vesículas que contienen estas enzimas se desprenden del aparato de Golgi. Dos enzimas son responsables de la fijación de la etiqueta de manosa-6-fosfato: La N-acetilglucosamina fosfotransferasa y la N-acetilglucosamina fosfoglucosidasa.
Esta vesícula, ahora en el citoplasma, se une a un endosoma tardío que es otro orgánulo ácido unido a la membrana. El endosoma tardío tiene bombas de protones dentro de su membrana que mantienen su entorno interno ácido. El bajo pH provoca la disociación de la proteína del receptor de manosa-6-fosfato. Este receptor puede entonces reciclarse de vuelta al aparato de Golgi.
El grupo fosfato también se elimina de la etiqueta de manosa-6-fosfato, para evitar que toda la proteína vuelva al aparato de Golgi. El endosoma tardío puede acabar madurando hasta convertirse en un lisosoma, después de haber recibido las enzimas del aparato de Golgi.
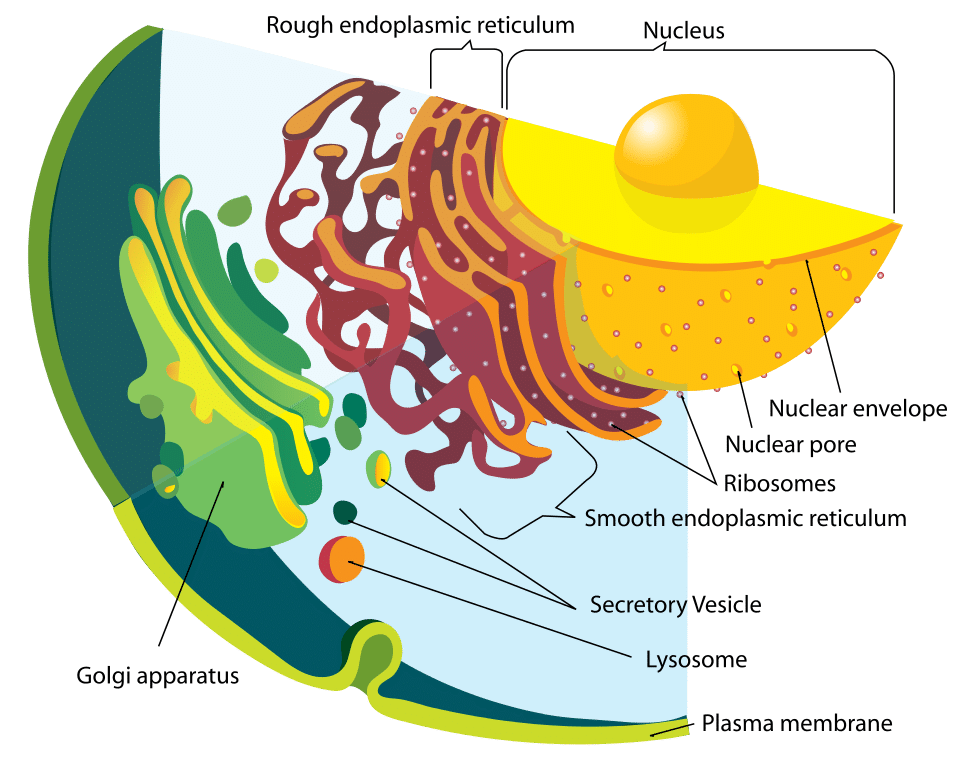 Figura 1 – Diagrama del sistema de endomembranas
Figura 1 – Diagrama del sistema de endomembranasFunción
Las enzimas hidrolíticas contenidas en el lisosoma permiten destruir las partículas extrañas. Los lisosomas desempeñan un papel importante en la fagocitosis. Cuando los macrófagos fagocitan partículas extrañas, las contienen dentro de un fagosoma. A continuación, el fagosoma se une a un lisosoma para formar un fagolisosoma.
Estas enzimas son fundamentales en los mecanismos de destrucción independientes del oxígeno. Los lisosomas también ayudan a defenderse de la entrada de patógenos a través de la endocitosis al degradar los patógenos antes de que lleguen al citoplasma.
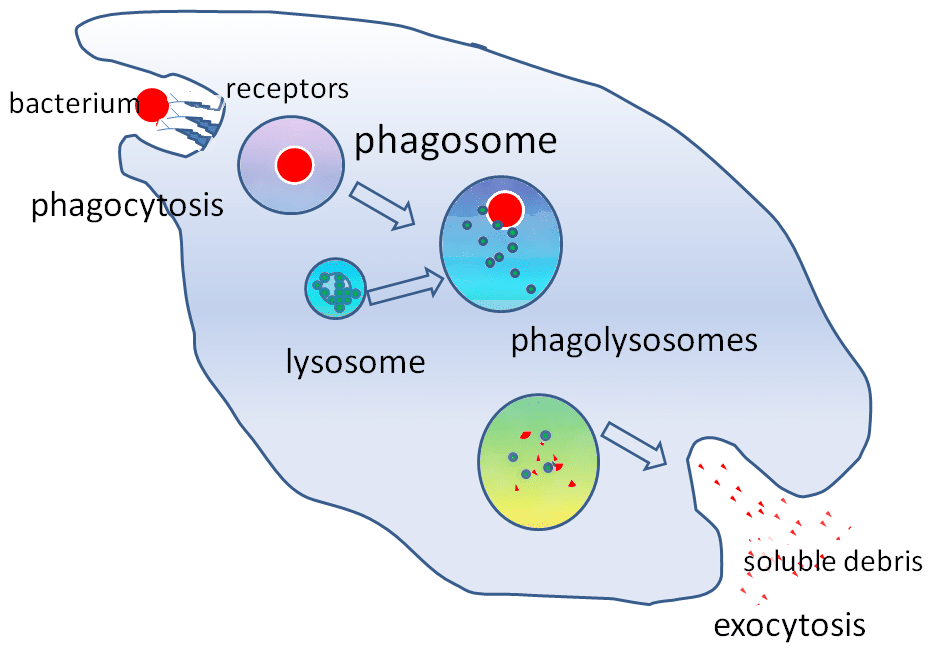 Figura 2 – El papel del lisosoma en la fagocitosis
Figura 2 – El papel del lisosoma en la fagocitosisRelevancia clínica
La enfermedad de las células I
Está causada por defectos genéticos en la enzima N-acetilglucosamina fosfotransferasa. Esta enzima es vital para la adición de manosa-6-fosfato a las proteínas dirigidas al lisosoma. Esto hace que las enzimas lisosomales no se dirijan correctamente. Como resultado, se encuentran cantidades significativas tanto en la orina como en el torrente sanguíneo.
Enfermedad de almacenamiento lisosómico
Son un grupo de condiciones genéticas que afectan a los lisosomas. La enfermedad varía mucho en cuanto a los signos, los síntomas y la demografía de los pacientes. Existen varias clasificaciones; la más común de estas afecciones es la enfermedad de Gaucher. Está causada por una deficiencia de la beta-glucocerebrosidasa. Esta enzima es necesaria para descomponer el glucocerebrósido. Sin esta enzima, el glucocerebrósido puede acumularse dentro de las células, lo que puede dañarlas. Los síntomas incluyen hepatoesplenomegalia y anemia.