
徴候・症状
第5染色体に関連するSMA(SMN関連)、0~4型
0~4型の脊髄性筋萎縮症(SMA)では、運動ニューロンという神経細胞の中にどれだけSMNタンパク質があるかによって症状が重いものから軽いものと連続的に変化します。 (SMNタンパク質が多いほど、症状が現れる時期が遅くなり、病気の経過も穏やかになります。 SMN1遺伝子の機能不全を補うSMN2遺伝子のコピー数が、SMAの重症度と発症年齢を決定します。
0型SMAの子どもの母親は、妊娠後期に胎動が少なくなり、出産が早くなることが報告されています。 これらの新生児は、重度の脱力感、低緊張、心臓障害を呈します。 新生児は運動面のマイルストーンを達成することができません。 多くの場合、これらの新生児は顔面神経麻痺、刺激に対する反応の欠如、先天性心臓障害を有しています1,2,3。 SMA0型と診断された患者は、生後6カ月までに、ときには生後1カ月以内に呼吸不全で死亡します。
出生時または出生直後に顕著なSMA症状を示した子どもは通常、非常に弱く、呼吸、吸引、飲み込みが困難で、発達のマイルストーンである一人座り(SMA1型またはウェルドニヒ・ホフマン病)には至らないことが多いのです。 SMA1型の子どもたちは、人工呼吸器や栄養チューブなどの技術により、呼吸や栄養を補助することで、何年も生き続けることができます。 研究の現状と新しい治療法に関する詳細は、「研究」または「MDA、小児患者における脊髄性筋萎縮症の治療にZolgensmaのFDA承認を祝う」をご覧ください。
SMA2型(別名Dubowitz病、または中間SMA)の症状は生後約3~15カ月の乳児に始まり、補助なしで座っていられるようになりますが、自立歩行することはありません。 この症状はSMA全体の約20%を占めます。 筋力低下は近位部(体の中心に近い部分)に多く、上肢よりも下肢に関与します。 4 呼吸器系の合併症が常に心配されますが、SMA2型の子どもは通常成人期まで生き、多くは長生きします。5
年長児や10代で筋力低下が始まり、立ったり歩いたりできるようになっても、人生の後半でその能力を失う場合、SMA3型(別名:軽症SMA、若年性発症、Kugelberg-Welander病)の疾患名で呼ばれることがあります。 SMA3型は、SMAの約30%を占めます。 3型は思春期に歩けなくなる人もいますが、成人になっても歩ける人もいます。 10代後半から成人期にかけて発症するSMAは、4型(晩発性SMA)と呼ばれます。 このタイプでは一般に寿命は正常です。 1型から4型では、体の中心に近い筋肉(近位筋)が、中心から遠い筋肉よりも影響を受けやすいか、少なくとも早く影響を受けるのが一般的です。 例えば、太ももの筋肉は下腿や足の筋肉よりも弱くなります。
脚は腕よりも先に弱くなる傾向があります。 手はいずれ弱くなるかもしれないが、通常最も強く保たれ、たとえ弱くなったとしても、通常コンピューターのキーボードや現代生活の他の基本的な機能をタイプするのに十分な強さを保つ。
SMAにおける最も深刻な危険は、呼吸に必要な筋肉の弱さから来る。 生涯を通じて呼吸機能に注意し、感染症に迅速に対応することが必要です。
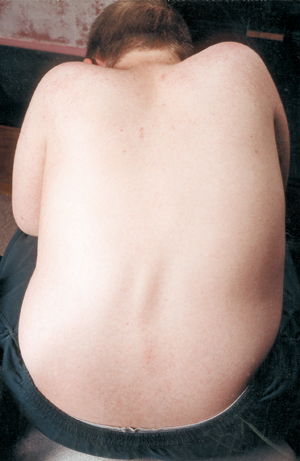
SMAのもう一つの合併症は脊椎の湾曲で、通常は側弯と呼ばれる左右に曲がるタイプです。 脊柱側弯症は、柔軟な柱である背骨を支える筋肉が弱くなることで起こります。 脊柱側弯症は、非常に不快で、体位や可動性を妨げ、子供(または大人)の身体イメージを損なう可能性があります。
SMAの子どもの多くは、人生の早い段階で脊柱側湾症を示し始め、手術に適した時期になるまで、装具で治療することがよくあります。 外科医は一般に、成長が完了するか、ほぼ完了するまで待ってから、脊椎を手術でまっすぐにし、癒合させたいと考えています。
第5染色体に関連しないSMA
SMAのいくつかの型は、第5染色体やSMN欠損症に関連しません。 これらの病型は、重症度や最も影響を受ける筋肉が大きく異なります。 5番染色体に関連した型のように、ほとんどの型が近位筋に影響を与える一方で、遠位筋に影響を与える型も存在し、少なくとも初期には体の中心から遠い筋肉に影響を与えます。 J. Med. Genet. (2008). doi:10.1136/jmg.2008.057950
.