
Les lysosomes sont des organites sphériques, liés à la membrane, qui sont générés par l’appareil de Golgi. Ils contiennent des enzymes hydrolytiques, et fonctionnent ainsi comme une partie du système de recyclage de la cellule.
Dans cet article, nous examinerons la structure, la synthèse et la fonction des lysosomes, et nous considérerons leur pertinence pour la pratique clinique.
Structure
Les lysosomes sont des organites acides liés à la membrane que l’on trouve dans les cellules, généralement d’environ 1 micromètre de longueur. Les lysosomes contiennent de nombreuses enzymes hydrolytiques qui catalysent les réactions d’hydrolyse.
La membrane entourant le lysosome est vitale pour s’assurer que ces enzymes ne fuient pas dans le cytoplasme et endommagent la cellule de l’intérieur. Afin de maintenir le pH acide du lysosome, des protons sont activement transportés dans l’organite à travers la membrane lysosomale.
Synthèse
Le lysosome et les enzymes qu’il contient sont synthétisés séparément. Les protéines lysosomales se forment de la même manière que n’importe quelle autre protéine. La première étape est l’initiation de la production de brins d’ARNm à partir de segments d’ADN pertinents. Les brins d’ARNm se dirigent vers le réticulum endoplasmique rugueux, où les ribosomes construisent les enzymes hydrolytiques.
Important, ceux-ci sont marqués avec du mannose-6-phosphate dans l’appareil de Golgi pour les cibler vers le lysosome. En conséquence, les vésicules contenant ces enzymes bourgeonnent de l’appareil de Golgi. Deux enzymes sont responsables de la fixation de l’étiquette mannose-6-phosphate : La N-acétylglucosamine phosphotransférase et la N-acétylglucosamine phosphoglycosidase.
Cette vésicule, maintenant dans le cytoplasme, se lie ensuite avec un endosome tardif qui est un autre organite acide, lié à la membrane. L’endosome tardif possède des pompes à protons dans sa membrane qui maintiennent son environnement interne acide. Le faible pH entraîne la dissociation de la protéine du récepteur du mannose-6-phosphate. Ce récepteur peut alors être recyclé vers l’appareil de Golgi.
Le groupe phosphate est également retiré de l’étiquette mannose-6-phosphate, afin d’empêcher la protéine entière de retourner vers l’appareil de Golgi. L’endosome tardif peut éventuellement mûrir en lysosome, après avoir reçu les enzymes de l’appareil de Golgi.
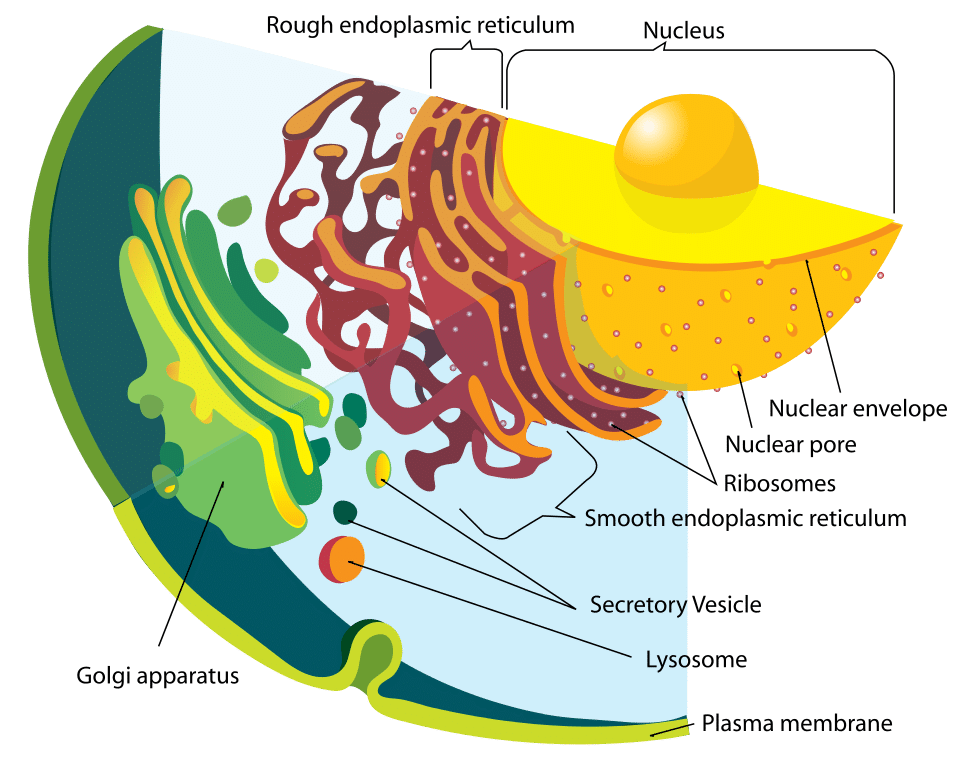 Figure 1 – Schéma du système endomembranaire
Figure 1 – Schéma du système endomembranaireFonction
Les enzymes hydrolytiques contenues dans le lysosome permettent de détruire les particules étrangères. Les lysosomes jouent un rôle important dans la phagocytose. Lorsque les macrophages phagocytent des particules étrangères, ils les contiennent dans un phagosome. Le phagosome va ensuite se lier à un lysosome pour former un phagolysosome.
Ces enzymes sont essentielles dans les mécanismes de destruction indépendants de l’oxygène. Les lysosomes aident également à se défendre contre l’entrée des pathogènes par endocytose en dégradant les pathogènes avant qu’ils n’atteignent le cytoplasme.
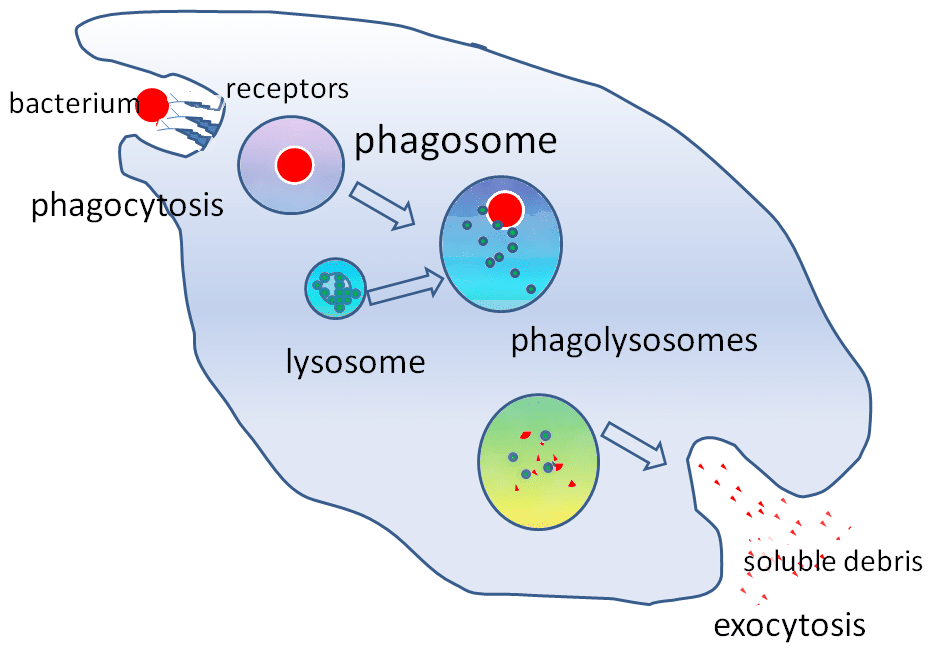 Figure 2 – Le rôle du lysosome dans la phagocytose
Figure 2 – Le rôle du lysosome dans la phagocytosePertinence clinique
Maladie des cellules I
Elle est causée par des défauts génétiques de l’enzyme N-acétylglucosamine phosphotransférase. Cette enzyme est essentielle pour l’ajout de mannose-6-phosphate aux protéines ciblées par le lysosome. Il en résulte que les enzymes lysosomales ne sont pas correctement ciblées. En conséquence, des quantités significatives sont trouvées à la fois dans l’urine et dans la circulation sanguine.
Lysosomal Storage Disease
Il s’agit d’un groupe de conditions génétiques affectant les lysosomes. Les signes, les symptômes et les caractéristiques démographiques des patients varient considérablement d’une affection à l’autre. Il existe plusieurs classifications ; la plus courante de ces affections est la maladie de Gaucher. Elle est causée par une déficience de la bêta-glucocérébrosidase. Cette enzyme est nécessaire pour décomposer le glucocérébroside. Sans cette enzyme, le glucocérébroside est capable de s’accumuler dans les cellules, ce qui peut les endommager. Les symptômes comprennent une hépatosplénomégalie et une anémie.