
Signes et symptômes
SMA liée au chromosome 5 (SMN-related), types 0-4
Dans l’atrophie musculaire spinale (SMA) de types 0 à 4, les symptômes varient sur un continuum de sévère à léger en fonction de la quantité de protéine SMN fonctionnelle présente dans les cellules nerveuses appelées motoneurones. (Plus la quantité de protéine SMN est importante, plus les symptômes apparaissent tard dans la vie et plus l’évolution de la maladie est légère. Le nombre de copies des gènes SMN2, qui peuvent compenser le mauvais fonctionnement des gènes SMN1, détermine la gravité de la SMA et l’âge d’apparition du patient.
Les mères d’enfants atteints de SMA de type 0 peuvent signaler une diminution des mouvements du fœtus en fin de grossesse et accoucher plus tôt. Ces nouveau-nés présentent une faiblesse sévère, une hypotonie et des malformations cardiaques. Les nouveau-nés n’atteignent aucun stade de développement moteur. Souvent, ces bébés présentent une diplégie faciale (paralysie faciale), une absence de réaction aux stimuli et une malformation cardiaque congénitale.1,2,3 Les patients atteints de SMA de type 0 meurent d’une insuffisance respiratoire avant l’âge de 6 mois et parfois dès le premier mois après la naissance.
Les enfants qui présentent des symptômes visibles de SMA à la naissance ou peu après sont généralement très faibles, ont des difficultés à respirer, à téter et à avaler et n’atteignent jamais le stade de développement où ils peuvent s’asseoir seuls (SMA de type 1 ou maladie de Werdnig-Hoffmann). Grâce à des technologies telles que la ventilation mécanique et les sondes d’alimentation pour aider à la respiration et à la nutrition, les enfants atteints de SMA de type 1 peuvent survivre pendant plusieurs années. Pour plus d’informations sur l’état actuel de la recherche et les nouveaux traitements, voir Recherche ou MDA célèbre l’approbation par la FDA du Zolgensma pour le traitement de l’amyotrophie spinale chez les patients pédiatriques.
Les symptômes de la SMA de type 2 (alias maladie de Dubowitz, ou SMA intermédiaire) commencent chez les bébés âgés d’environ 3 à 15 mois qui apprennent à s’asseoir sans aide mais ne se tiennent pas debout ou ne marchent pas de manière indépendante. Cette présentation représente environ 20 % de tous les cas de SMA. La faiblesse musculaire est principalement proximale (proche du centre du corps) et concerne davantage les membres inférieurs que les membres supérieurs. Habituellement, les muscles du visage et des yeux ne sont pas affectés.4 Bien que les complications respiratoires soient une menace constante, les enfants atteints de SMA de type 2 vivent généralement jusqu’au début de l’âge adulte, et beaucoup vivent plus longtemps.5
Lorsque la faiblesse musculaire commence chez les enfants plus âgés et les adolescents qui apprennent à se tenir debout et à marcher mais perdent ces capacités plus tard dans la vie, la maladie peut être étiquetée SMA de type 3 (alias SMA légère, apparition juvénile ou maladie de Kugelberg-Welander). La SMA de type 3 représente environ 30 % des cas de SMA. Bien que certains patients atteints du type 3 cessent de marcher à l’adolescence, d’autres marchent jusqu’à l’âge adulte. La plupart des patients développent des déformations des pieds, une scoliose et une faiblesse des muscles respiratoires.
La SMA qui se manifeste à la fin de l’adolescence ou à l’âge adulte est appelée type 4, ou SMA à déclenchement tardif. La durée de vie est généralement normale dans ce type. Les patients sont capables d’atteindre toutes les étapes motrices et de maintenir leur déambulation tout au long de leur vie.4
Dans les SMA de types 1 à 4, les muscles les plus proches du centre du corps (muscles proximaux) sont généralement plus affectés, ou du moins affectés beaucoup plus tôt, que les muscles plus éloignés du centre. Par exemple, les muscles des cuisses sont plus faibles que les muscles du bas des jambes et des pieds.
Les jambes ont tendance à s’affaiblir avant les bras. Les mains peuvent finir par s’affaiblir, mais elles restent généralement les plus fortes le plus longtemps et, même si elles s’affaiblissent, elles restent généralement assez fortes pour taper sur un clavier d’ordinateur et d’autres fonctions de base de la vie moderne.
Le danger le plus grave dans la SMA vient de la faiblesse des muscles nécessaires à la respiration. Une attention particulière à la fonction respiratoire est nécessaire tout au long de la vie, avec une attention rapide aux infections. Votre médecin peut vous aider avec les détails du maintien de la santé respiratoire, y compris l’élimination des sécrétions et peut-être la ventilation assistée (pas nécessairement 24 heures sur 24).
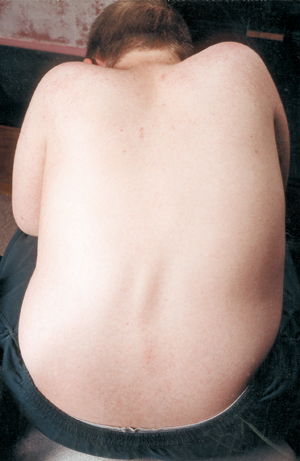
Une autre complication médicale de la SMA est la courbure de la colonne vertébrale, généralement un type de courbure d’un côté à l’autre appelé scoliose. La scoliose se produit en raison de la faiblesse des muscles qui soutiennent normalement la colonne vertébrale, qui est une colonne flexible. La scoliose peut être très inconfortable, interférer avec la position et la mobilité, et nuire à l’image corporelle de l’enfant (ou de l’adulte). Certaines études ont montré que les courbures de la colonne vertébrale, si elles sont sévères, peuvent interférer avec la respiration.
De nombreux enfants atteints de SMA commencent à montrer une courbe scoliotique tôt dans la vie, qui est souvent traitée avec une orthèse jusqu’à ce que le bon moment pour la chirurgie soit atteint. Les chirurgiens aiment généralement attendre que la croissance soit terminée ou presque avant de redresser et de fusionner chirurgicalement la colonne vertébrale. Ils tiennent également compte de la fonction pulmonaire de l’enfant et de la vitesse à laquelle la courbure de la colonne vertébrale est susceptible de progresser.
La SMA n’est pas liée au chromosome 5
Certaines formes de SMA ne sont pas liées au chromosome 5 ou au déficit en SMN. Ces formes varient beaucoup en termes de gravité et de muscles les plus touchés. Alors que la plupart des formes, comme celle liée au chromosome 5, affectent surtout les muscles proximaux, il existe d’autres formes qui affectent surtout les muscles distaux, ceux qui sont plus éloignés du centre du corps, du moins au début.
- Rudnik-Schöneborn, S. et al. La cardiopathie congénitale est une caractéristique de l’amyotrophie spinale infantile sévère. J. Med. Genet. (2008). doi:10.1136/jmg.2008.057950
- Grotto, S. et al. Type 0 Spinal Muscular Atrophy : Further Delineation of Prenatal and Postnatal Features in 16 Patients. J. Neuromuscul. Dis. (2016). doi:10.3233/JND-160177
- Menke, L. A. et al. Défauts cardiaques congénitaux dans l’amyotrophie spinale de type I : Un rapport clinique de deux frères et sœurs et une revue de la littérature. Am. J. Med. Genet. Part A (2008). doi:10.1002/ajmg.a.32233
- Butchbach, M. E. R. Copy Number Variations in the Survival Motor Neuron Genes : Implications pour l’amyotrophie spinale et d’autres maladies neurodégénératives. Front. Mol. Biosci. (2016). doi:10.3389/fmolb.2016.00007
- Zerres, K. & Schöneborn, S. R. Histoire naturelle dans l’amyotrophie spinale proximale : Analyse clinique de 445 patients et suggestions pour une modification des classifications existantes. Arch. Neurol. (1995). doi:10.1001/archneur.19900540290108025