
Anzeichen und Symptome
SMA im Zusammenhang mit Chromosom 5 (SMN-bezogen), Typen 0-4
Bei der spinalen Muskelatrophie (SMA) der Typen 0 bis 4 variieren die Symptome auf einem Kontinuum von schwer bis leicht, je nachdem, wie viel funktionsfähiges SMN-Protein in den Nervenzellen, den Motoneuronen, vorhanden ist. („SMN“ steht für „survival of motor neuron“). Je mehr SMN-Protein vorhanden ist, desto später im Leben beginnen die Symptome und desto milder ist der Krankheitsverlauf. Die Anzahl der Kopien der SMN2-Gene, die eine Fehlfunktion der SMN1-Gene ausgleichen können, bestimmt den Schweregrad der SMA und das Alter des Patienten.
Mütter von Kindern, die von SMA Typ 0 betroffen sind, können in der späten Schwangerschaft eine Abnahme der fötalen Bewegungen feststellen und früher entbinden. Diese Neugeborenen weisen schwere Schwäche, Hypotonie und Herzfehler auf. Die Neugeborenen erreichen keine motorischen Meilensteine. Häufig haben diese Babys eine Gesichtslähmung, eine mangelnde Reaktion auf Reize und einen angeborenen Herzfehler.1,2,3 Patienten, bei denen SMA Typ 0 diagnostiziert wird, sterben bis zum Alter von 6 Monaten an Atemversagen, manchmal sogar schon innerhalb des ersten Monats nach der Geburt.
Kinder, die bereits bei oder kurz nach der Geburt deutliche SMA-Symptome aufweisen, sind in der Regel sehr schwach, haben Schwierigkeiten beim Atmen, Saugen und Schlucken und erreichen nie den Entwicklungsschritt, dass sie in der Lage sind, selbständig zu sitzen (SMA Typ 1 oder Werdnig-Hoffmann-Krankheit). Mit Hilfe von Technologien wie mechanischer Beatmung und Ernährungssonden zur Unterstützung von Atmung und Ernährung können Kinder mit SMA Typ 1 einige Jahre überleben. Weitere Informationen über den aktuellen Stand der Forschung und neue Behandlungsmöglichkeiten finden Sie unter Forschung oder MDA feiert FDA-Zulassung von Zolgensma zur Behandlung der spinalen Muskelatrophie bei pädiatrischen Patienten.
SMA Typ 2 (auch bekannt als Dubowitz-Krankheit oder intermediäre SMA): Die ersten Symptome treten bei Säuglingen im Alter von etwa 3 bis 15 Monaten auf, die lernen, ohne Hilfe zu sitzen, aber noch nicht selbstständig stehen oder gehen können. Diese Form macht etwa 20 % aller SMA-Fälle aus. Die Muskelschwäche ist überwiegend proximal (nahe der Körpermitte) und betrifft eher die unteren als die oberen Gliedmaßen. Normalerweise sind die Gesichts- und Augenmuskeln nicht betroffen.4 Obwohl Komplikationen bei der Atmung eine ständige Bedrohung darstellen, leben Kinder mit SMA Typ 2 in der Regel bis ins junge Erwachsenenalter, viele sogar länger.5
Wenn die Muskelschwäche bei älteren Kindern und Jugendlichen beginnt, die stehen und gehen lernen, diese Fähigkeiten aber später im Leben verlieren, kann die Krankheit als SMA Typ 3 (auch bekannt als milde SMA, juveniler Beginn oder Kugelberg-Welander-Krankheit) bezeichnet werden. SMA Typ 3 macht etwa 30 % der Fälle von SMA aus. Einige Patienten mit Typ 3 hören im Jugendalter auf zu laufen, andere gehen bis ins Erwachsenenalter. Die meisten Patienten entwickeln Fußdeformitäten, Skoliose und Atemmuskelschwäche.
SMA, die im späten Teenager- oder Erwachsenenalter auftritt, wird als Typ 4 oder spät einsetzende SMA bezeichnet. Bei diesem Typ ist die Lebenserwartung im Allgemeinen normal. Die Patienten sind in der Lage, alle motorischen Meilensteine zu erreichen und können ihr ganzes Leben lang gehen.4
Bei den SMA-Typen 1 bis 4 sind die Muskeln, die sich näher an der Körpermitte befinden (proximale Muskeln), in der Regel stärker oder zumindest viel früher betroffen als die Muskeln, die weiter vom Zentrum entfernt sind. Zum Beispiel sind die Muskeln der Oberschenkel schwächer als die Muskeln der Unterschenkel und Füße.
Die Beine werden in der Regel vor den Armen schwächer. Die Hände können mit der Zeit schwächer werden, aber sie bleiben in der Regel am längsten stark, und selbst wenn sie schwächer werden, bleiben sie in der Regel stark genug für das Tippen auf einer Computertastatur und andere grundlegende Funktionen des modernen Lebens.
Die größte Gefahr bei SMA geht von der Schwäche der für die Atmung notwendigen Muskeln aus. Die Atmungsfunktion muss während des gesamten Lebens sorgfältig überwacht werden, und Infektionen müssen rechtzeitig erkannt werden. Ihr Arzt kann Ihnen bei den Einzelheiten der Gesunderhaltung der Atemwege behilflich sein, einschließlich der Entfernung von Sekreten und eventuell unterstützter Beatmung (nicht unbedingt rund um die Uhr).
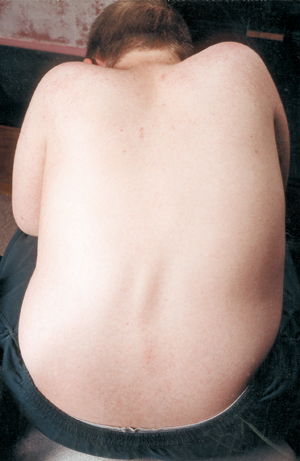
Eine weitere medizinische Komplikation bei SMA ist die Wirbelsäulenverkrümmung, in der Regel eine seitliche Verkrümmung, die Skoliose genannt wird. Die Skoliose entsteht durch eine Schwäche der Muskeln, die normalerweise die Wirbelsäule stützen, die eine flexible Säule ist. Skoliose kann sehr unangenehm sein, die Haltung und Beweglichkeit beeinträchtigen und das Körperbild eines Kindes (oder Erwachsenen) schädigen. Einige Studien haben gezeigt, dass starke Wirbelsäulenverkrümmungen die Atmung beeinträchtigen können.
Viele Kinder mit SMA zeigen schon früh im Leben eine skoliotische Verkrümmung, die oft mit einem Korsett behandelt wird, bis der richtige Zeitpunkt für eine Operation erreicht ist. Chirurgen warten in der Regel gerne, bis das Wachstum abgeschlossen oder fast abgeschlossen ist, bevor sie die Wirbelsäule operativ begradigen und fixieren. Sie berücksichtigen auch die Lungenfunktion des Kindes und wie schnell die Wirbelsäulenkrümmung wahrscheinlich fortschreitet.
SMA nicht mit Chromosom 5 verbunden
Einige Formen von SMA sind nicht mit Chromosom 5 oder SMN-Mangel verbunden. Diese Formen unterscheiden sich stark im Schweregrad und in den am stärksten betroffenen Muskeln. Während die meisten Formen, wie die mit Chromosom 5 zusammenhängende Form, vor allem die proximalen Muskeln betreffen, gibt es andere Formen, bei denen vor allem die distalen Muskeln betroffen sind, die weiter vom Körperzentrum entfernt sind, zumindest zu Beginn.
- Rudnik-Schöneborn, S. et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J. Med. Genet. (2008). doi:10.1136/jmg.2008.057950
- Grotto, S. et al. Type 0 Spinal Muscular Atrophy: Further Delineation of Prenatal and Postnatal Features in 16 Patients. J. Neuromuscul. Dis. (2016). doi:10.3233/JND-160177
- Menke, L. A. et al. Congenital heart defects in spinal muscular atrophy type I: Ein klinischer Bericht von zwei Geschwistern und eine Übersicht über die Literatur. Am. J. Med. Genet. Part A (2008). doi:10.1002/ajmg.a.32233
- Butchbach, M. E. R. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front. Mol. Biosci. (2016). doi:10.3389/fmolb.2016.00007
- Zerres, K. & Schöneborn, S. R. Natural History in Proximal Spinal Muscular Atrophy: Klinische Analyse von 445 Patienten und Vorschläge für eine Modifikation bestehender Klassifikationen. Arch. Neurol. (1995). doi:10.1001/archneur.19900540290108025