
Lysosomen sind kugelförmige, membrangebundene Organellen, die vom Golgi-Apparat gebildet werden. Sie enthalten hydrolytische Enzyme und fungieren so als Teil des Recyclingsystems der Zelle.
In diesem Artikel werden wir uns mit der Struktur, Synthese und Funktion von Lysosomen befassen und ihre Bedeutung für die klinische Praxis betrachten.
Struktur
Lysosomen sind saure, membrangebundene Organellen, die sich in den Zellen befinden und in der Regel etwa 1 Mikrometer lang sind. Lysosomen enthalten zahlreiche hydrolytische Enzyme, die Hydrolysereaktionen katalysieren.
Die Membran, die das Lysosom umgibt, ist wichtig, damit diese Enzyme nicht in das Zytoplasma austreten und die Zelle von innen heraus schädigen. Um den sauren pH-Wert des Lysosoms aufrechtzuerhalten, werden Protonen aktiv durch die lysosomale Membran in das Organell transportiert.
Synthese
Das Lysosom und die darin enthaltenen Enzyme werden separat synthetisiert. Lysosomale Proteine werden auf die gleiche Weise gebildet wie jedes andere Protein. Der erste Schritt ist die Initiierung der mRNA-Strangproduktion aus den entsprechenden DNA-Abschnitten. Die mRNA-Stränge wandern zum rauen endoplasmatischen Retikulum, wo Ribosomen die hydrolytischen Enzyme aufbauen.
Wichtig ist, dass diese im Golgi-Apparat mit Mannose-6-Phosphat markiert werden, um sie zum Lysosom zu bringen. Die Vesikel, die diese Enzyme enthalten, lösen sich daraufhin vom Golgi-Apparat. Zwei Enzyme sind für die Anbringung der Mannose-6-Phosphat-Marke verantwortlich: N-Acetylglucosamin-Phosphotransferase und N-Acetylglucosamin-Phosphoglykosidase.
Dieses Vesikel, das sich nun im Zytoplasma befindet, verbindet sich dann mit einem späten Endosom, einer weiteren sauren, membrangebundenen Organelle. Das späte Endosom hat Protonenpumpen in seiner Membran, die seine innere Umgebung sauer halten. Der niedrige pH-Wert bewirkt die Dissoziation des Proteins vom Mannose-6-Phosphat-Rezeptor. Dieser Rezeptor kann dann in den Golgi-Apparat zurückgeführt werden.
Die Phosphatgruppe wird auch von der Mannose-6-Phosphat-Marke entfernt, um zu verhindern, dass das gesamte Protein in den Golgi-Apparat zurückkehrt. Das späte Endosom kann schließlich zu einem Lysosom heranreifen, nachdem es die Enzyme aus dem Golgi-Apparat erhalten hat.
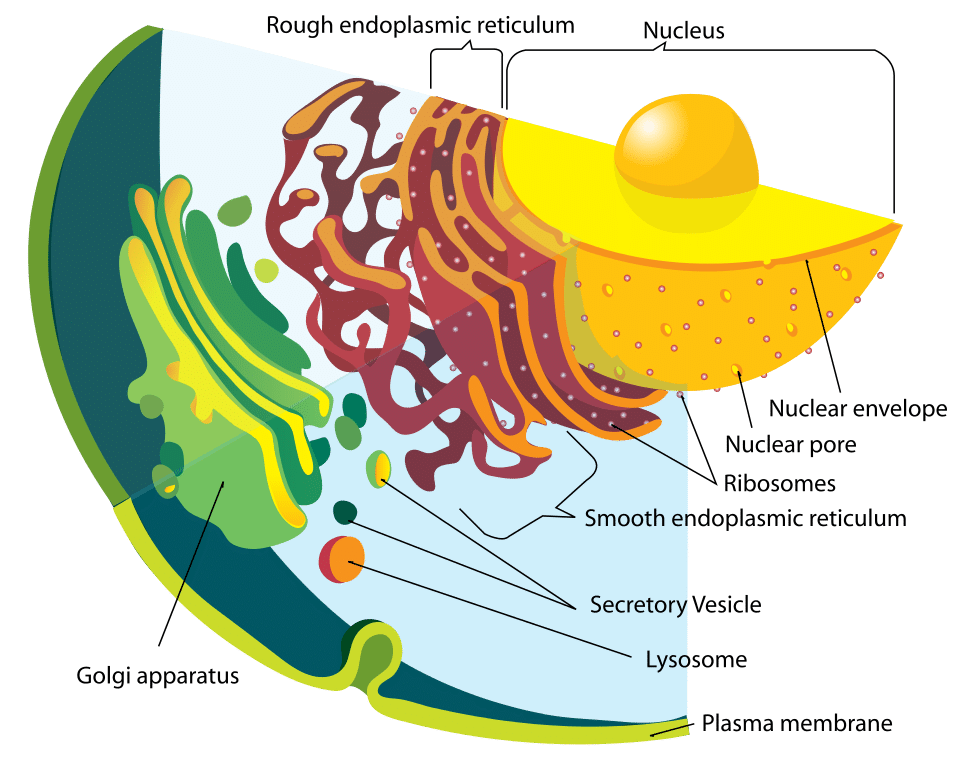 Abbildung 1 – Schema des Endomembransystems
Abbildung 1 – Schema des EndomembransystemsFunktion
Die im Lysosom enthaltenen hydrolytischen Enzyme ermöglichen die Zerstörung von Fremdpartikeln. Lysosomen spielen eine wichtige Rolle bei der Phagozytose. Wenn Makrophagen fremde Partikel phagozytieren, schließen sie sie in einem Phagosom ein. Das Phagosom verbindet sich dann mit einem Lysosom, um ein Phagolysosom zu bilden.
Diese Enzyme sind entscheidend für sauerstoffunabhängige Abtötungsmechanismen. Lysosomen tragen auch dazu bei, das Eindringen von Krankheitserregern durch Endozytose zu verhindern, indem sie Krankheitserreger abbauen, bevor sie das Zytoplasma erreichen.
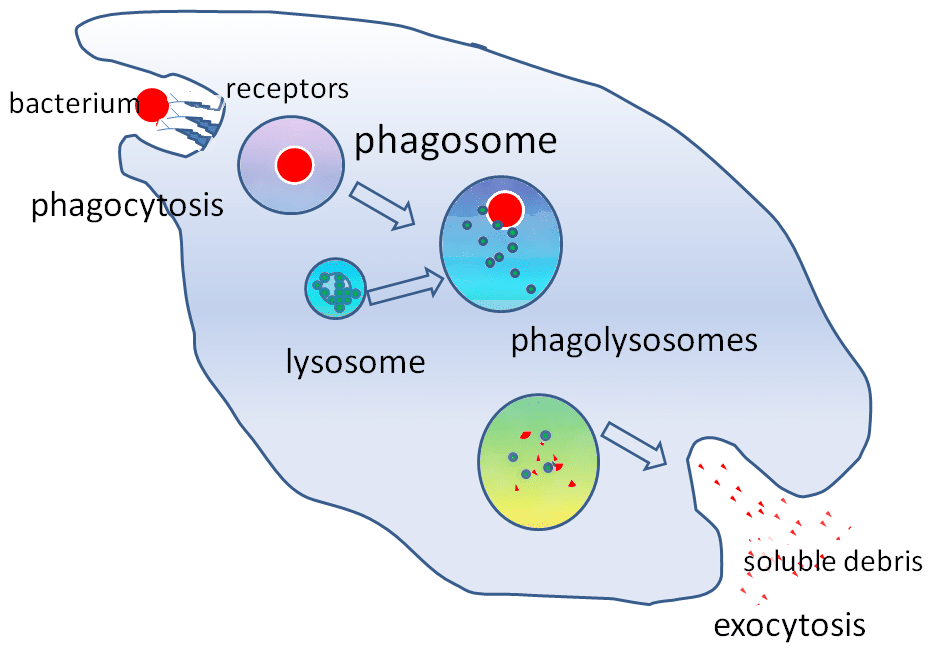 Abbildung 2 – Die Rolle der Lysosomen bei der Phagozytose
Abbildung 2 – Die Rolle der Lysosomen bei der PhagozytoseKlinische Relevanz
I-Zell-Krankheit
Diese wird durch genetische Defekte des Enzyms N-Acetylglucosamin-Phosphotransferase verursacht. Dieses Enzym ist entscheidend für die Anlagerung von Mannose-6-Phosphat an lysosomale Proteine. Dies führt dazu, dass die lysosomalen Enzyme nicht richtig angegriffen werden. Infolgedessen werden erhebliche Mengen sowohl im Urin als auch im Blut gefunden.
Lysosomale Speicherkrankheit
Dies ist eine Gruppe von genetischen Erkrankungen, die die Lysosomen betreffen. Diese Erkrankungen unterscheiden sich stark in ihren Anzeichen, Symptomen und demografischen Merkmalen der Patienten. Es gibt mehrere Klassifizierungen; die häufigste dieser Erkrankungen ist die Gaucher-Krankheit. Sie wird durch einen Mangel an beta-Glukozerebrosidase verursacht. Dieses Enzym ist für den Abbau von Glukozerebrosid erforderlich. Ohne dieses Enzym kann sich das Glukozerebrosid in den Zellen anreichern, was diese schädigen kann. Zu den Symptomen gehören Hepatosplenomegalie und Anämie.