
Signos y síntomas
Amia ligada al cromosoma 5 (relacionada con el SMN), tipos 0-4
En la atrofia muscular espinal (AME) de los tipos 0 a 4, los síntomas varían en un continuo que va de grave a leve según la cantidad de proteína SMN funcional que haya en las células nerviosas llamadas neuronas motoras. («SMN» significa supervivencia de la neurona motora.) Cuanta más proteína SMN haya, más tarde empezarán los síntomas y más leve será el curso de la enfermedad. El número de copias de los genes SMN2, que pueden compensar el mal funcionamiento de los genes SMN1, determina la gravedad de la AME y la edad de inicio del paciente.
Las madres de niños afectados por la AME tipo 0 pueden informar de una disminución del movimiento fetal al final del embarazo y dar a luz antes. Estos recién nacidos se presentan con debilidad grave, hipotonía y defectos cardíacos. Los recién nacidos no alcanzan ningún hito motor. A menudo, estos bebés presentan diplejía facial (parálisis facial), falta de reacción a los estímulos y un defecto cardíaco congénito.1,2,3 Los pacientes diagnosticados con AME tipo 0 mueren de insuficiencia respiratoria a los 6 meses de edad y, a veces, en el primer mes después del nacimiento.
Los niños que presentan síntomas notables de AME en el momento del nacimiento o poco después suelen ser muy débiles; tienen dificultades para respirar, succionar y tragar; y nunca alcanzan el hito del desarrollo de poder sentarse por sí mismos (AME tipo 1 o enfermedad de Werdnig-Hoffmann). Con tecnología como la ventilación mecánica y las sondas de alimentación para ayudar a la respiración y la nutrición, los niños con AME tipo 1 pueden sobrevivir durante varios años. Para más información sobre el estado actual de la investigación y los nuevos tratamientos, consulte Investigación o La MDA celebra la aprobación por la FDA de Zolgensma para el tratamiento de la atrofia muscular espinal en pacientes pediátricos.
Los síntomas de la AME tipo 2 (también conocida como enfermedad de Dubowitz, o AME intermedia) comienzan en bebés de aproximadamente 3 a 15 meses de edad que aprenden a sentarse sin ayuda pero no se ponen de pie ni caminan de forma independiente. Esta presentación representa alrededor del 20% de todos los casos de AME. La debilidad muscular es predominantemente proximal (cerca del centro del cuerpo) y afecta más a las extremidades inferiores que a las superiores. Por lo general, los músculos de la cara y los ojos no se ven afectados.4 Aunque las complicaciones respiratorias son una amenaza constante, los niños con AME tipo 2 suelen vivir hasta la edad adulta, y muchos viven más tiempo.5
Cuando la debilidad muscular comienza en niños mayores y adolescentes que aprenden a ponerse de pie y a caminar, pero pierden estas capacidades más adelante, la enfermedad puede ser etiquetada como AME tipo 3 (también conocida como AME leve, aparición juvenil o enfermedad de Kugelberg-Welander). La AME tipo 3 representa alrededor del 30% de los casos de AME. Aunque algunos enfermos del tipo 3 dejan de caminar en la adolescencia, otros caminan hasta bien entrada la edad adulta. La mayoría de los pacientes desarrollan deformidades en los pies, escoliosis y debilidad de los músculos respiratorios.
La AME que aparece al final de la adolescencia o en la edad adulta se llama tipo 4, o AME de inicio tardío. La duración de la vida es generalmente normal en este tipo. Los pacientes son capaces de alcanzar todos los hitos motores y mantener su deambulación durante toda la vida.4
En los tipos de AME 1 a 4, los músculos más cercanos al centro del cuerpo (músculos proximales) suelen estar más afectados, o al menos se afectan mucho antes, que los músculos más alejados del centro. Por ejemplo, los músculos de los muslos son más débiles que los de la parte inferior de las piernas y los pies.
Las piernas tienden a debilitarse antes que los brazos. Las manos pueden debilitarse con el tiempo, pero suelen ser las más fuertes y, aunque se debiliten, suelen seguir siendo lo suficientemente fuertes como para escribir en un teclado de ordenador y otras funciones básicas de la vida moderna.
El peligro más grave en la AME proviene de la debilidad de los músculos necesarios para respirar. Es necesario prestar una cuidadosa atención a la función respiratoria a lo largo de toda la vida, con una pronta atención a las infecciones. Su médico puede ayudarle con los detalles del mantenimiento de la salud respiratoria, incluida la eliminación de las secreciones y quizás la ventilación asistida (no necesariamente las 24 horas del día).
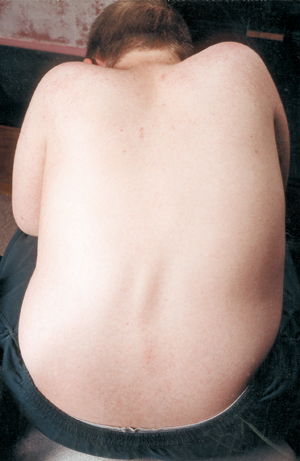
Otra complicación médica de la AME es la curvatura de la columna vertebral, normalmente un tipo de curvatura de lado a lado llamada escoliosis. La escoliosis se produce por la debilidad de los músculos que normalmente sostienen la columna vertebral, que es una columna flexible. La escoliosis puede ser muy incómoda, interferir con la posición y la movilidad, y dañar la imagen corporal del niño (o del adulto). Algunos estudios han demostrado que las curvaturas de la columna vertebral, si son graves, pueden interferir con la respiración.
Muchos niños con AME comienzan a mostrar una curva escoliótica a principios de la vida, que a menudo se trata con un corsé hasta que llega el momento adecuado para la cirugía. Por lo general, los cirujanos prefieren esperar hasta que el crecimiento esté completo o casi completo antes de enderezar y fusionar quirúrgicamente la columna vertebral. También tienen en cuenta la función pulmonar del niño y la rapidez con la que puede progresar la curvatura de la columna.
La AME no está vinculada al cromosoma 5
Algunas formas de AME no están vinculadas al cromosoma 5 o a la deficiencia de SMN. Estas formas varían mucho en gravedad y en los músculos más afectados. Mientras que la mayoría de las formas, como la relacionada con el cromosoma 5, afectan sobre todo a los músculos proximales, existen otras formas que afectan sobre todo a los músculos distales, los más alejados del centro del cuerpo, al menos al principio.
- Rudnik-Schöneborn, S. et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J. Med. Genet. (2008). doi:10.1136/jmg.2008.057950
- Grotto, S. et al. Type 0 Spinal Muscular Atrophy: Delineación adicional de las características prenatales y postnatales en 16 pacientes. J. Neuromuscul. Dis. (2016). doi:10.3233/JND-160177
- Menke, L. A. et al. Congenital heart defects in spinal muscular atrophy type I: Un informe clínico de dos hermanos y una revisión de la literatura. Am. J. Med. Genet. Part A (2008). doi:10.1002/ajmg.a.32233
- Butchbach, M. E. R. Copy Number Variations in the Survival Motor Neuron Genes: Implicaciones para la atrofia muscular espinal y otras enfermedades neurodegenerativas. Front. Mol. Biosci. (2016). doi:10.3389/fmolb.2016.00007
- Zerres, K. & Schöneborn, S. R. Natural History in Proximal Spinal Muscular Atrophy: Análisis clínico de 445 pacientes y sugerencias para una modificación de las clasificaciones existentes. Arch. Neurol. (1995). doi:10.1001/archneur.19900540290108025